
近十几年来,肠道微生物逐渐成为国自然的重点资助方向之一。因为样本
相对容易获取
,研究门槛较低,同时可以通过高通量测序快速上手,实现“
短、平、快
”发表文章。
今天馆长介绍的主角是肠道微生物,不过这类文章已经相对“泛滥”,想再蹭这类国自然热点发高分,无论是数据上还是实验方法上都要“更上一层楼”才行。所以今天馆长给大家分享一波新套路:与传统的肠道微生物宏基因组研究不同,还融汇了
机器学习
,以及利用
孟德尔随机化
来分析因果关系。更多精彩下面咱们就一起来看看:
1、大规模多人群队列研究
:这是
首次
对来自中国、日本和意大利的八个长寿人群进行的大规模集成宏基因组队列研究,涉及1156份粪便样本,
为研究提供了丰富的跨地区数据
。通过对大量粪便样本进行宏基因组测序,提供了肠道微生物群落组成的全面视图,
这在长寿研究中是相对较新的研究方法。
2、肠道微生物与长寿的因果关系
:通过孟德尔随机化(MR)分析,不仅识别了与长寿相关的肠道微生物物种和途径,还进一步推断了它们与长寿之间的潜在因果关系,
这是以往研究中较少涉及的
。
不得不说孟德尔随机化真的是“万金油”般的存在,大家赶快行动起来吧!掌握思路,复现思路,发文就差这关键的一步!欢迎扫下方二维码,寻找更多创新思路!


题目
:长寿人群肠道微生物组的一致性特征
杂志
:Gut Microbes
影响因子
:IF=12.2
发表时间
:2024年08月28日
后台回复“
666
”获取原文献,编号20240904
研究背景
近年来,对百岁老人肠道微生物群的研究受到了广泛关注。一些研究已经描述了意大利、日本和中国百岁老人的肠道微生物群特征,提供了与衰老相关的肠道微生物组成的潜在见解。例如,Akkermansia 菌在百岁老人的肠道中富集,而 Bacteroides 和 Faecalibacterium 在健康的年轻对照组中更丰富。然而,目前对于特定微生物、功能途径或微生物代谢产物与长寿或衰老相关的病理生理机制的了解还相对有限。
研究思路
研究者通过整合来自不同地区的长寿人群的粪便样本,利用宏基因组测序技术深入分析了肠道微生物的物种水平功能,并结合孟德尔随机化来探索肠道微生物群与长寿之间的潜在因果关系。
数据来源
|
数据集
|
数据库
|
数据类型
|
详细信息
|
|
PRJNA675598
|
GEO
|
宏基因组
|
C1(日本):330个样本,包括176个百岁老人或非百岁老人(C组)、110个较年轻老年人(E组)和44个年轻人(Y组)。
|
|
PRJNA613947
|
GEO
|
宏基因组
|
C2(中国启东县):348个样本
|
|
PRJEB25514
|
ENA
|
宏基因组
|
C3(意大利撒丁岛):59个样本
|
|
PRJNA553191
|
GEO
|
宏基因组
|
C4(意大利罗马尼亚地区):62个样本
|
|
PRJNA624763
|
GEO
|
宏基因组
|
C5(中国如皋市):30个样本
|
|
PRJNA772518
|
GEO
|
宏基因组
|
C6(中国都江堰):86个样本
|
|
CNP0002519
|
CNGBdb
|
宏基因组
|
C7(中国海南):25个样本
|
|
CNP0004699
|
CNGBdb
|
宏基因组
|
C8(中国梅州队列):216个样本,包括35个百岁老人(C组)、142个较年轻老年人(E组)和39个年轻人(Y组)
|
主要结果
1.
八个长寿种群肠道菌群的多样性和组成
研究者通过对来自八个长寿人群的1156份粪便样本进行分析(图1a),发现长寿个体(C组)与较年轻老年人(E组)和年轻人(Y组)相比,
具有更高的肠道微生物多样性
。
α-多样性分析
显示C组和E组的肠道微生物多样性高于Y组,但当将E组和Y组合并为非长寿组时,与长寿组相比没有显著差异。此外还发现
338种微生物是所有年龄组共有的核心成员
,而C组独有39种物种,E组独有34种,Y组独有25种物种。
β-多样性分析
揭示了六个队列中不同年龄组间肠道微生物组成的显著差异。在
属水平
上,长寿人群的肠道微生物群中主要优势属包括Bacteroides、Phocaeicola、Bifidobacterium等,而非长寿人群则以Phocaeicola、Prevotella、Faecalibacterium等为优势属(图1b)。
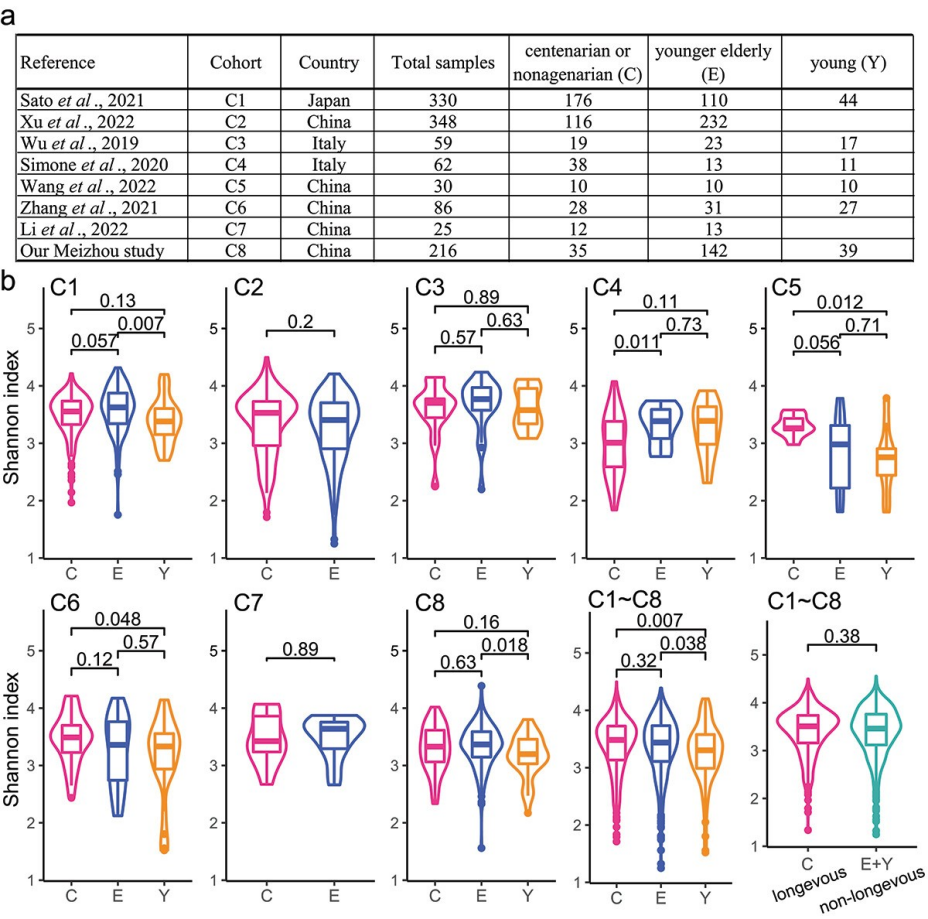
图1 8个长寿种群粪便样品信息及不同年龄组粪便样品微生物α多样性
2.
八个长寿种群肠道微生物群内显著改变的物种
在对八个长寿人群的肠道微生物群研究中,发现了与长寿相关的
六种显著改变的菌种
:Eisenbergiella tayi、Methanobrevibacter smithii、Hungatella hathewayi、Ruthenibacterium lactatiformans、Enterocloster lavalensis和Faecalibacterium prausnitzii(图2a)。
E. tayi
在七个数据集中富集(图2b),而其他四种在至少六个数据集中富集(图2c-d)。
F. prausnitzii
在六个数据集中减少。此外,
Akkermansia muciniphila
在三个最大的队列中富集。这些菌种可能是与长寿相关的肠道微生物标志。
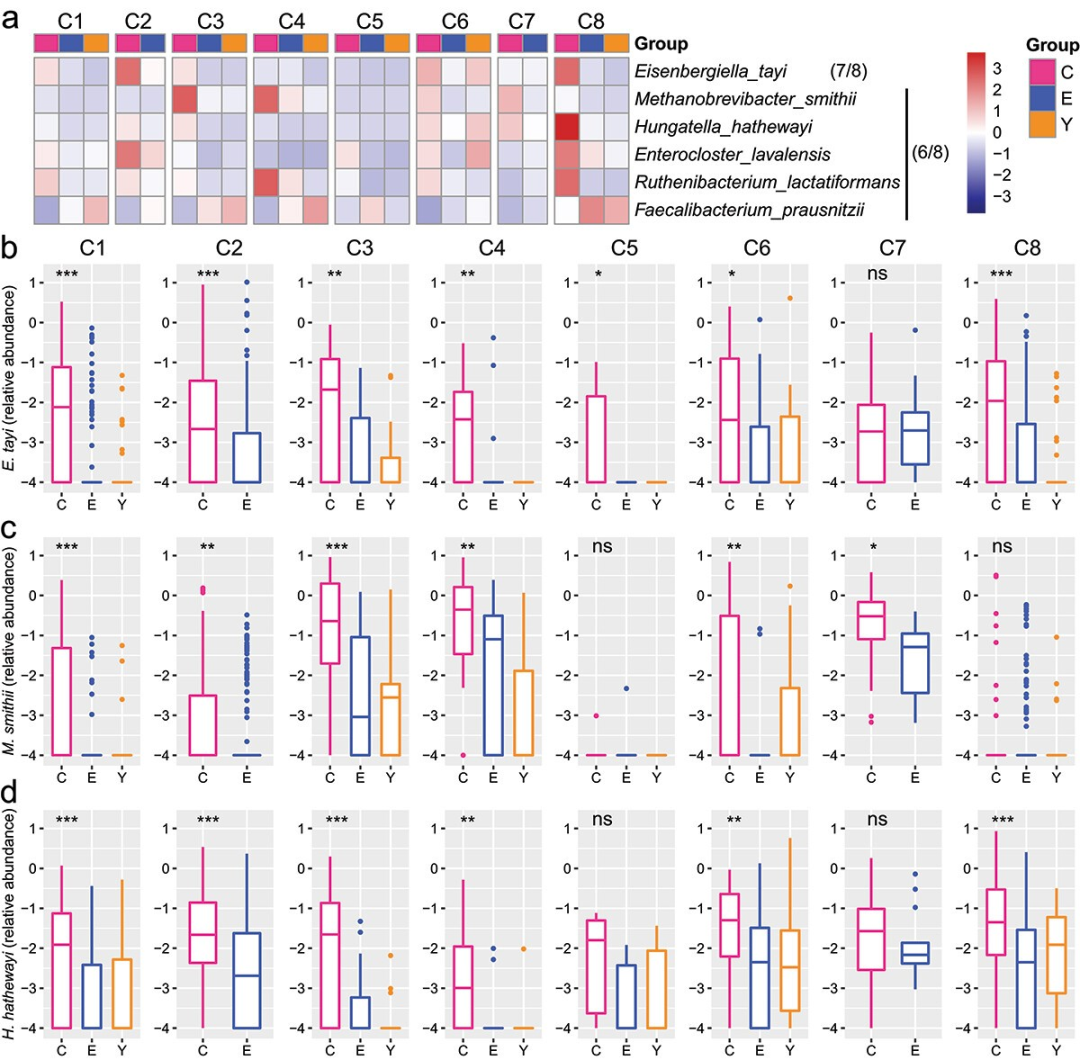
图2 八个长寿组群中不同年龄组之间相对丰度变化最一致的物种
3.
长寿人群肠道微生物群生物标志物
研究者利用
随机森林分类器
评估了肠道微生物种类是否能作为区分长寿人群与非长寿人群的潜在生物标志物。在C1至C7数据集(发现队列)中,AUC为0.86,在C8数据集(验证队列)中,AUC为0.79(图3a-b)。准确性最高的10种微生物包括Neglecta timonensis、Desulfovibrio fairfieldensis等(图3c),这些在所有数据集中均富集于长寿人群。当考虑所有八个数据集时,AUC达到0.85(图3d),其中6种微生物与之前发现一致(图3e)。
这些结果表明,肠道微生物种类有潜力作为长寿的生物标志物
。
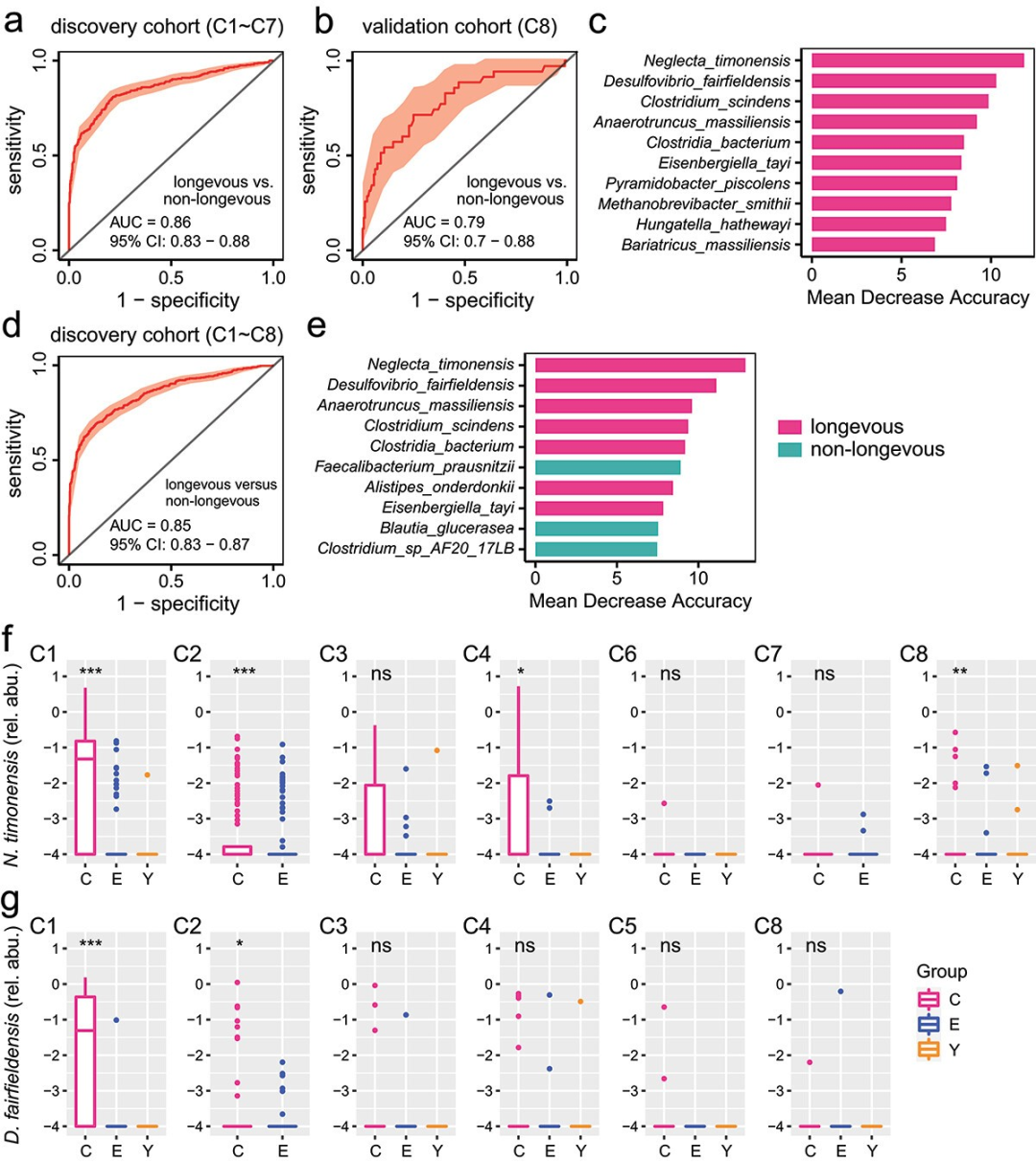
图3 采用随机森林(RF)分类器选择物种标记,区分长寿个体和非长寿个体
4.
在长寿种群中与不同物种相关的微生物途径的改变
研究者进一步探讨了这些种类所涉及的
微生物途径
。Eisenbergiella tayi在PWY-7031和PWY-6749途径中贡献显著(图4a-b),且这些途径在长寿组中的相对丰度更高。Hungatella hathewayi在嘌呤核苷降解I(P164-PWY)途径中也有显著贡献(图4c-d)。Desulfovibrio fairfieldensis在1,4-二羟基-6-萘甲酸生物合成I(PWY-7374)途径中显著富集(图4e-f)。Methanobrevibacter smithii参与了九个在长寿组中富集的途径,包括产甲烷作用。
这些发现揭示了与长寿相关的肠道微生物途径,可能对健康长寿有重要影响
(图4h)。
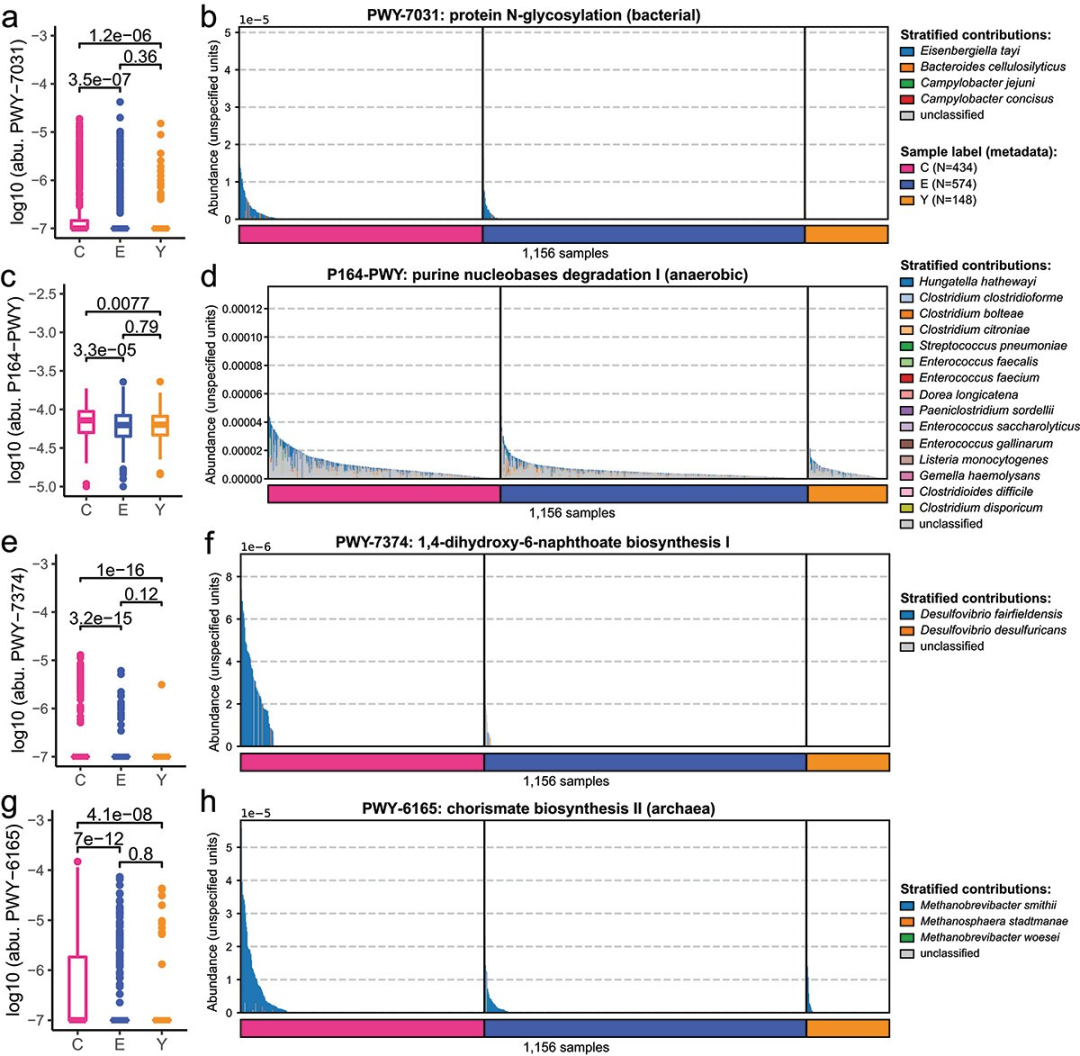
图
4 C
,
E
和
Y
组中四个关键微生物途径的相对丰度
进一步探讨了与长寿相关的微生物途径中涉及的关键酶及其产生菌种。在PWY-7031中(图5a),鉴定了核心生物合成酶pglC、pglA、pglJ、pglH和pglI,而在PWY-6749途径中,主要酶包括legB、legC、legH、legG、legI和legF。这些酶在长寿组中的相对丰度显著高于其他年龄组(图5b)。Eisenbergiella tayi和Bacteroides eggerthii在pglH酶的产生中起主导作用(图5c),而E. tayi和Bacteroides cellulosilyticus对EC 2.7.7.82酶的贡献显著(图5d)。这些发现表明
E. tayi通过特定酶参与了PWY-7031和PWY-6749途径
。
此外,Hungatella hathewayi在P164-PWY途径中通过甘氨酸还原酶(EC 1.21.4.2)和乙酰激酶(EC 2.7.2.1)显著贡献(图5e),这些酶将甘氨酸转化为乙酰磷酸,进而转化为乙酸。Desulfovibrio fairfieldensis在PWY-7374途径中通过chorismate脱水酶(EC 4.2.1.151)和futalosine水解酶(EC 3.2.2.26)显著贡献(图5f)。
这些酶在长寿组样本中的丰度显著高于其他年龄组
,而
这些菌种可能通过特定途径影响长寿
。
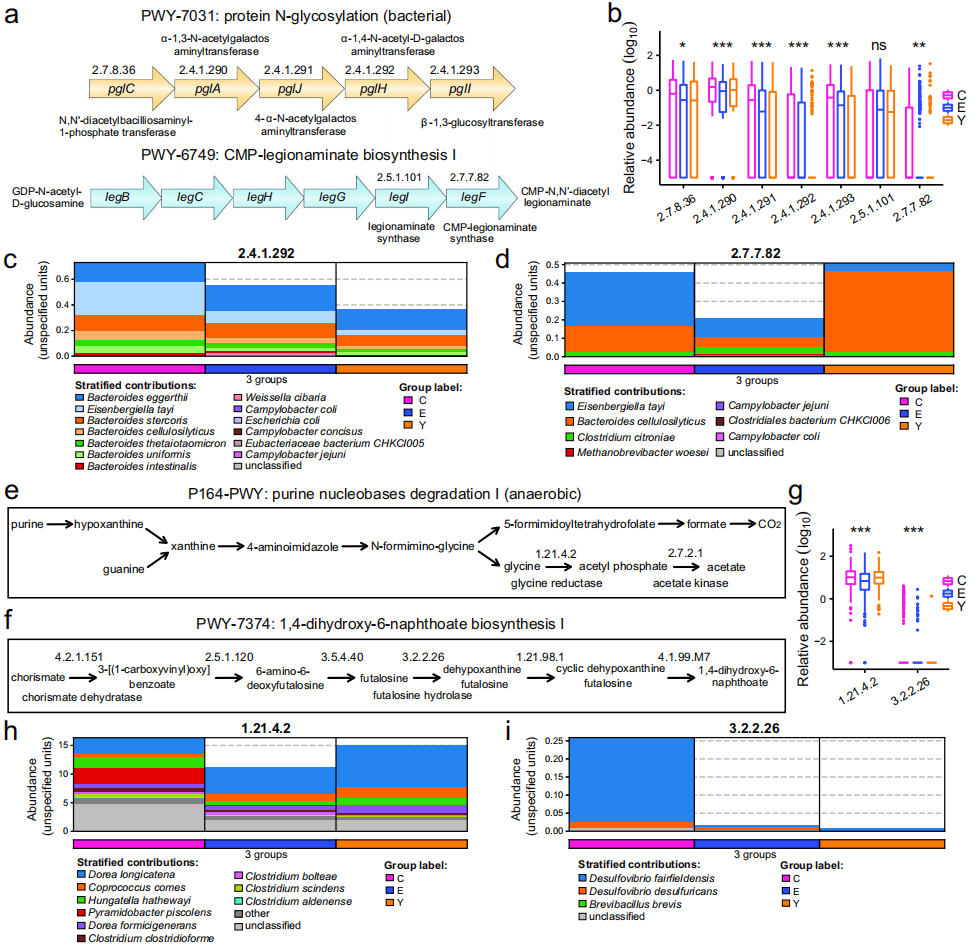
图5 几个关键的微生物途径及其相关酶










