


摘 要:目的 制备载姜黄素两亲性星状聚酯纳米粒(Cur-NPs),以解决其稳定性差、生物利用度低等问题。方法 通过开环聚合反应和酯化反应合成两亲性星状聚酯(DPE-PCL-mPEG)作为纳米粒的载体材料,傅立叶变换显微红外光谱(FT-IR)、1H-NMR和凝胶渗透色谱(GPC)表征确定其结构和相对分子质量。溶剂挥发法制备Cur-NPs,考察其粒径、ξ电位、载药量、包封率。对Cur-NPs进行稳定性、体外释放、材料安全性、体外抗肿瘤和细胞摄取能力考察。结果 成功合成DPE-PCL-mPEG,制备的Cur-NPs平均粒径为(86.00±2.01)nm,ξ电位为(−9.40±0.09)mV,包封率为(95.51±1.23)%,载药量为(5.52±0.54)%。Cur-NPs具有良好的稳定性和缓释能力。细胞毒性、细胞摄取和体外抗肿瘤实验表明,空白纳米粒(blank-NPs)具有良好的生物安全性;相对于姜黄素溶液,Cur-NPs对人胶质瘤U251细胞的生长抑制作用更明显,并且具有更强的入胞能力。结论 Cur-NPs理化性质理想,能有效提高药物体外生物活性,为姜黄素的临床应用提供了新的解决方案。
目前,癌症是全球第2大致死疾病,每年超过1000万人被确诊为癌症[1],90%以上的癌症在潜伏期没有明显症状,直到中晚期才被发现。化疗是临床上治疗中晚期癌症的主要方式之一,可以延缓癌症的扩散和转移;但缺乏靶向性,对正常组织有明显的毒副作用[2],容易导致多药耐药和变态反应。姜黄素(curcumin,Cur)是从姜黄根茎中提取的天然产物[3],经FDA认证具有很好的安全性,可以通过多种分子机制抑制肿瘤细胞的增殖、分化、侵袭和转移[4-5],还可以逆转多药耐药性[5]。但姜黄素难溶于水、易降解、体内生物利用度低等特点[6]严重限制其临床应用。因此,急需研究新剂型以解决药物递送的一系列问题。近年来,随着纳米技术[7-8]的发展,纳米给药体系可以有效地改变药物在体内的药动学特征,从而实现药物的缓释及靶向递送,减轻毒副作用、提高疗效[9-11]。星状聚合物因其具有独特的空间形态,自组装成粒后,内部可形成巨大的空腔;相比直链聚合物,星状聚合物制备的纳米粒具有更高的载药量和包封率[12-13],是一种良好的纳米载体。聚乙二醇是广泛使用的亲水性材料[14],常与其他载体材料共价连接[15-17],以增加纳米粒的稳定性和体内循环时间[17]。结合星状聚合物与聚乙二醇特性,将两者通过共价键连接成为一种新型两亲性星状聚酯,不仅可以实现姜黄素的高效包载,还可以有效避免网状内皮系统对纳米粒的清除,延长体内循环时间。此外,纳米粒可以通过实体瘤的高通透性和滞留效应(EPR效应),使药物在肿瘤部位富集,增加药物进入肿瘤细胞几率,从而提高药物生物利用度实现治疗肿瘤目的。基于此,本实验选用双季戊四醇(dipentaerythritol,DPE)、ɛ-环己内酯(ɛ-cyclocaprolactone,ɛ-CL)和聚乙二醇单甲醚(polyethylene glycolmonomethyl ether,mPEG,相对分子质量5000)通过开环聚合和酯化反应合成两亲性星状聚酯DPE-PCL-mPEG,以姜黄素为模型药物,采用溶剂挥发法[18]制备载姜黄素纳米粒(Cur- NPs),对其物理化学性质进行表征。选取人胶质瘤U251细胞为模型细胞,进一步考察该纳米粒的细胞毒性、体外抗肿瘤活性和入胞能力。以期得到一种粒径小、安全性高、稳定性好、抗肿瘤活性良好的纳米给药体系,为姜黄素的临床应用提供新的解决途径。1 材料与仪器
1.1 材料
姜黄素,质量分数98%,杭州广林生物医药有限公司;异辛酸亚锡[Sn(Oct)2],化学纯,国药集团化学试剂有限公司;ɛ-CL,分析纯,阿拉丁公司;DPE、mPEG、N,N′-二环已基碳二亚胺(N,N′-dicyclohexylcarbodiimide,DCC)、4-二甲氨基吡啶(4-dimethylaminopyridine,DMAP)、聚山梨酯80(化学纯),阿拉丁公司;丁二酸酐、三乙胺,上海凌峰化学试剂有限公司;四氢呋喃、乙腈为色谱纯;人胶质瘤U251细胞,中国科学院细胞库;DMEM培养基、胎牛血清,浙江天杭生物科技股份有限公司;0.25%胰蛋白酶,Gibco公司;96孔板、细胞培养皿,耐思生物科技有限公司;MTT,索莱宝科技有限公司。1.2 仪器
Malvern ZS90激光纳米粒径测定仪,英国马尔文仪器有限公司;UV-2102紫外可见分光光度计,美国尤尼柯公司;LGJ-10冷冻干燥机,杭州诺丁科学器材有限公司;傅立叶变换显微红外光谱仪(FT- IR)、XIR高速冷冻离心机、超净台,Thermo Fisher公司;IL-161CT二氧化碳培养箱,施都凯仪器设备上海有限公司;酶标仪,Biotek公司;荧光显微镜,Olympus公司;核磁共振波谱仪,Bruker公司;X射线衍射仪,PNAlytical公司;透射电镜,日本电子株式会社。2 方法与结果
2.1 DPE-PCL和DPE-PCL-mPEG的合成
DPE-PCL-mPEG合成分成3个步骤:首先,通过开环聚合反应生成DPE-PCL。其次,用丁二酸酐修饰mPEG生成mPEG-COOH。最后,通过酯化反应,生成DPE-PCL-mPEG。具体步骤如图1所示。
2.1.1 DPE-PCL的合成 1 mmol的DPE、60 mmol的ɛ-CL和0.06 mmol的Sn(Oct)2(催化剂)置于50 mL的三口瓶中,N2保护下,450 r/min磁力搅拌,120 ℃反应24 h。反应结束后,冷却至室温,加入10 mL二氯甲烷溶解,再逐滴加至冰乙醚中,沉淀纯化,抽滤收集粗产物。粗产物经二次沉淀纯化后于真空干燥箱干燥至恒定质量,称定质量计算DPE- PCL产率。
2.1.2 mPEG-COOH的合成 2 mmol mPEG、3 mmol的丁二酸酐和15 mL的吡啶在100 mL三口瓶内搅拌溶解。加入0.002 mmol DMAP(催化剂)和0.02 mmol三乙胺(缚酸剂),N2保护下,450 r/min磁力搅拌,室温反应24 h。反应结束后,逐滴加至冰乙醚中,沉淀纯化,抽滤收集粗产物。10 mL二氯甲烷溶解粗产物后逐滴加至冰乙醚中,再次沉淀纯化,抽滤收集产物,于真空干燥箱干燥至恒定质量,称定质量计算mPEG-COOH产率。
2.1.3 DPE-PCL-mPEG的合成 称取1 mmol的DPE-PCL和1 mmol的mPEG-COOH于50 mL的三口瓶中,加入1 mL乙腈搅拌溶解。再加入1 mmol的DCC(催化剂)和0.1 mmol的DMAP(催化剂),N2保护下,450 r/min磁力搅拌,室温反应48 h。反应结束后收集产物,方法同“2.1.2”项。
2.2 DPE-PCL和DPE-PCL-mPEG的表征
通过FT-IR和1H-NMR确定DPE-PCL和DPE- PCL-mPEG结构;凝胶渗透色谱(GPC)[16]和1H- NMR分别计算DPE-PCL和DPE-PCL-mPEG相对分子质量。DPE-PCL的FT-IR结果如图2所示,其酯键中C=O的伸缩振动峰出现在1 726.3 cm−1,-C-O-C-的不对称振动峰和对称振动峰分别出现在1 295.5、1 191.5 cm−1。而2 945.3、2 866.3、1 471.0、732.5 cm−1这4个吸收峰归属于PCL片段上的亚甲基中C-H的特征峰。DPE-PCL的1H-NMR分析结果如图3-A所示。图中-O-CH2-、-CH2-CH2-CH2-CH2-CH2-、-CH2- CH2-CH2-CH2-CH2-和-CH2-COO-均为PCL片段上的特征峰,其化学位移分别为δ 2.32(a)、1.39(b)、1.66(d)、4.06(c)。而PCL的末端亚甲基质子峰(-CH2-OH),由于受末端羟基的影响其化学位移向低场移至δ 3.65(e)。此外其相对分子质量可以通过c与e之间的峰面积积分比计算,结果见表1。FT-IR和1H-NMR的结果表明DPE-PCL已成功合成。
DPE-PCL-mPEG的FT-IR结果如图2所示,与DPE-PCL的FT-IR结果相比较,DPE-PCL-mPEG除了PCL片段中出现的酯键和亚甲基的特征峰,还出现了mPEG中醚键(-C-O-C-)的特征峰,分别位于1 140.0、1 061.7 cm−1。DPE-PCL-mPEG的1H- NMR分析结果如图3-B所示,δ 3.65(g)、3.39(f)为mPEG中-CH2-CH2O-和-OCH3的特征峰。而δ 2.32(a)、1.39(b)、1.66(d)、4.07(c)为DPE-PCL- mPEG中PCL片段中亚甲基的特征峰。此外,通过f与g之间的峰面积积分比计算其相对分子质量,结果见表1。FT-IR和1H-NMR的结果表明已成功合成DPE-PCL-mPEG。

DPE-PCL和DPE-PCL-mPEG的GPC分析结果如图4和表1所示,2个化合物的GPC曲线均是单峰且基本对称,多分散指数(PDI)分别为1.08和1.14,相对分子质量分布均比较集中。另外GPC中测得的数均相对分子质量与1H-NMR中计算得到的相近,表明该聚合物聚合度较均匀。
2.3 姜黄素分析方法的建立
2.3.1 色谱条件 色谱柱为Axxlaim® Polar Advantage II-C18柱(250 mm×4.6 mm,5 μm);流动相为乙腈-0.6%乙酸水溶液(60∶40);体积流量1.0 mL/min;进样量10 μL;检测波长420 nm;柱温30 ℃。
2.3.2 对照品溶液制备 精密称取姜黄素对照品2.0 mg,用乙腈溶解并定容至10 mL,配制成姜黄素对照品储备液。
2.3.3 专属性实验 取适量的姜黄素和纳米粒,乙腈溶解,配制成姜黄素溶液、空白纳米粒溶液(blank-NPs)、载药纳米粒溶液,0.22 μm微孔滤膜滤过,按“2.3.1”项色谱条件进样检测。HPLC检测图谱如图5所示,姜黄素保留时间为6.9 min,峰形较好,姜黄素与杂峰分离较好,载体材料对检测无干扰,方法专属性好,适用于纳米粒的包封率和载药量测定。

2.3.4 线性关系考察 取适量对照品溶液,用流动相稀释成质量浓度分别为0.1、0.5、1.0、2.0、4.0、6.0、8.0 μg/mL的系列对照品溶液,0.22 μm微孔滤过,检测姜黄素含量。以姜黄素的质量浓度为横坐标(X),峰面积积分值为纵坐标(Y)绘制标准曲线,进行线性回归,得到回归方程Y=1.566 9 X+0.008 6,R2=0.999 8,结果表明姜黄素在0.1~8.0 μg/mL线性关系良好,该方法适用于姜黄素的含量测定。
2.3.5 精密度试验 在线性范围内,精密量取对照品储备液,分别配制质量浓度为0.5、2.0、6.0 μg/mL的对照品溶液,HPLC测定质量浓度并计算RSD分别为0.10%、0.20%、0.07%、RSD均<2%,该方法精密度良好,方法可行。
2.3.6 回收率试验 分别取质量浓度为6.0 μg/mL的姜黄素对照品溶液2、3、4 mL置于10 mL量瓶中,取0.5 mL空白纳米粒溶液加入样品中,流动相定容,制得质量浓度分别为1.2、1.8、2.4 μg/mL的溶液,0.22 μm微孔滤过,按“2.3.1”项色谱条件进样检测,每个质量浓度平行操作3次,计算回收率。回收率试验结果显示,1.2、1.8、2.4 μg/mL的回收率分别为98.56%、99.25%、99.12%,RSD分别为1.79%、1.21%、0.98%,回收率均在95%~105%,RSD均<2%,符合方法学要求。
2.4 Cur-NPs的制备
2.4.1 制备方法 溶剂挥发法制备Cur-NPs混悬液,精密称取姜黄素和DPE-PCL-mPEG,丙酮溶解后缓慢滴加入纯水中,450 r/min磁力搅拌30 min。40 ℃下真空干燥3 h,4 ℃下低速离心10 min (6000 r/min)以除去游离姜黄素,得到Cur-NPs。空白纳米粒(blank-NPs)的制备除不加药物,其余步骤相同。
2.4.2 包封率和载药量考察 取Cur-NPs在15 000 r/min的转速下高速离心60 min,收集并干燥沉淀,精密称定质量,加丙酮溶解定容,测定姜黄素含量,根据公式计算纳米粒的载药量和包封率。
包封率=W姜黄素/W投药量
载药量=W姜黄素/WCur-NPs
W姜黄素为Cur-NPs中姜黄素的质量,W投药量为投入姜黄素的质量,WCur-NPs为Cur-NPs的质量
2.4.3 单因素考察 分别考察姜黄素与DPE-PCL- mPEG的质量比(药材比,1∶10、1∶15、1∶20、1∶25、1∶30)、有机相与水相的体积比(脂水比,1∶3、1∶4、1∶5、1∶6、1∶7)及姜黄素的质量浓度(1.5、2.0、2.5、3.0、3.5 mg/mL)这3个因素对制备Cur-NPs的影响。以平均粒径、PDI、包封率和载药量为指标,优化制备Cur-NPs的处方。
单因素考察结果见表2,姜黄素与DPE-PCL- mPEG的质量比、姜黄素的质量浓度、有机相与水相的体积比对纳米粒的大小、载药量和包封率均有影响,对PDI几乎没有影响。其中在姜黄素与DPE-PCL-mPEG的质量比为1∶20,有机相与水相的体积比为1∶5,姜黄素的质量浓度为2.5 mg/mL的条件下,Cur-NPs的平均粒径理想,载药量和包封率最高。
2.4.4 Box-Behnken效应面优化Cur-NPs处方 使用软件Design Expert 10中的Box-Behnken效应面,设置因素X1(姜黄素与DPE-PCL-mPEG的质量比)、X2(有机相与水相的体积比)和X3(姜黄素的质量浓度),输入相应的平均粒径、PDI、包封率和载药量。根据Hassan方法将粒径和PDI结果用公式dmax处理,包封率和载药量结果用公式dmin处理。最后根据公式计算总评归一值(OD),优化制备处方。
dmax=(Yi-Ymin)/(Ymax-Ymin)
dmin=(Ymin-Yi)/(Ymax-Ymin)
OD=(d1d2…dk)1/k
根据单因素结果确定X1、X2和X3 3个因素的考察范围分别为X1 1∶10~1∶30、X2 1∶3~1∶7和X3 1.5~3.5,每个因素设置5个水平,OD值和方差分析结果见表3、4,通过拟合得到方程:OD= 0.825 7-0.082 10 X1-0.017 85 X2+0.073 58 X3+0.030 73 X1X2+0.059 43 X1X3-0.038 83 X2X3- 0.029 26 X12-0.223 50 X22-0.563 40 X32。其三维曲线图见图6。最终得到最优处方:姜黄素与DPE- PCL-mPEG的质量比为1∶17.8,有机相与水相的体积比为1∶4.9,姜黄素的质量浓度为2.5 mg/mL。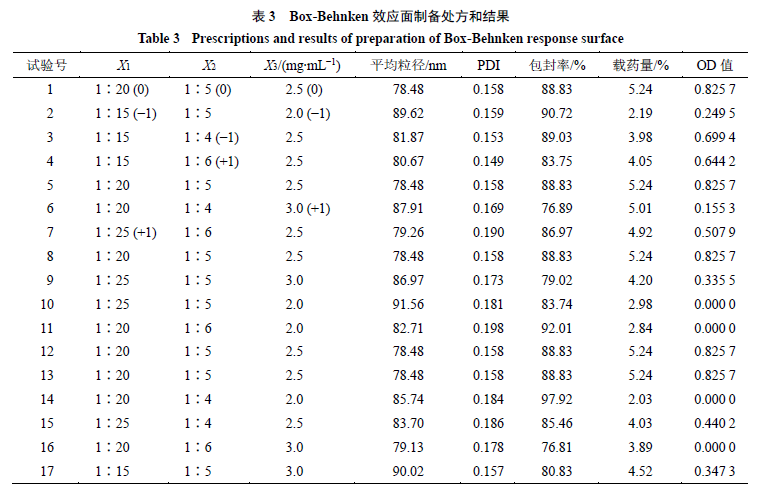
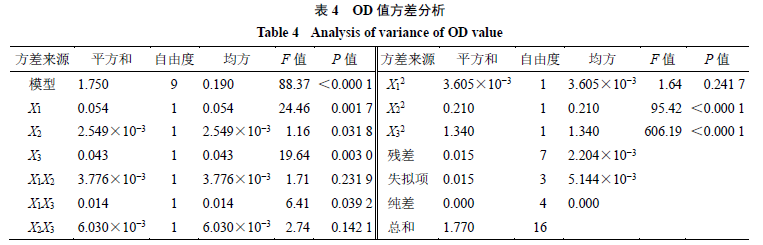
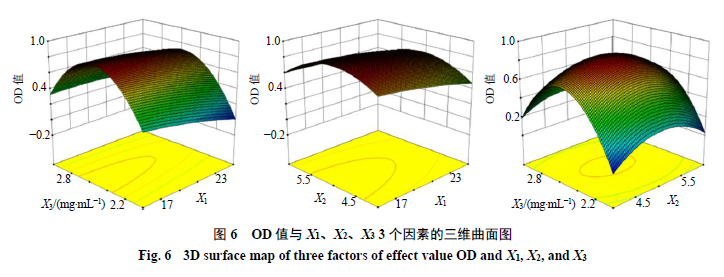
将Box-Behnken效应面中得到的处方进行验证,结果如表5所示,实验结果以表示。各指标实际值与预测值接近,表明该回归方程的建立具有统计学意义。Cur-NPs的平均粒径为(86.00±2.01)nm,可以通过EPR效应进入肿瘤区域[19]。另 外Cur-NPs的PDI小于0.2,表明纳米粒的粒径分布较窄,均一性良好。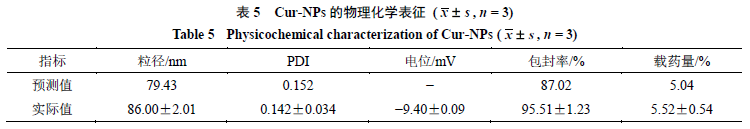
2.5 Cur-NPs的表征
2.5.1 纳米粒的平均粒径、ξ电位、包封率和载药量 取Cur-NPs,在25 ℃下,马尔文粒径仪测定其平均粒径和ξ电位;包封率和载药量计算方法同“2.4.2”项。结果见表5,Cur-NPs呈负电荷,可有效减少血浆蛋白在粒子表面吸附[19]和被网状内皮系统清除[20],增加其体内稳定性和循环能力;Cur- NPs具有较理想的包封率和载药量。
2.5.2 Cur-NPs的形态 取1滴blank-NPs或Cur- NPs置于铜网(200目)上,2%磷钨酸钠染色,滤纸吸去过量的染色剂,将样品在室温下干燥,在透射电子显微镜下观察纳米粒形态(图7),blank-NPs和Cur-NPs呈较规整的球形,呈单分散状态,平均粒径约为80 nm与激光粒度仪测定结果相符。

2.6 Cur-NPs的稳定性考察
取2 mL Cur-NPs分别加入8 mL 0.01 mol/L磷酸盐缓冲溶液(PBS,pH 7.4)和含10%血清(FBS)的0.01 mol/L PBS(pH 7.4),37 ℃下静置保存,在0、1、4、8、12、24 h取样并测定其平均粒径和ξ电位。Cur-NPs在PBS(pH 7.4)和PBS+10% FBS(pH 7.4)中的稳定性结果如图8所示。24 h内Cur- NPs在PBS中平均粒径从86.5 nm增大至88 nm,没有明显波动;但在PBS+10% FBS中粒径从86.5 nm增大至93 nm,可能是由于血清中的蛋白在Cur- NPs表面少量附着导致粒径略有增大,但10 h后粒径不再有明显的波动,表明Cur-NPs处于稳定状态。
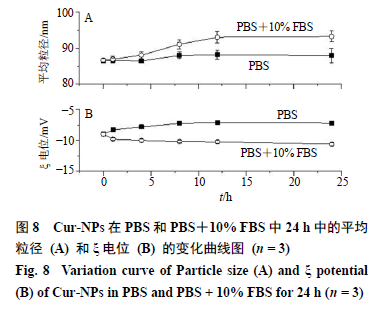
此外,Cur-NPs在PBS和PBS+10% FBS中ξ电位变化均在1 mV以内,没有明显改变。结果表明Cur- NPs稳定性良好。2.7 X-射线衍射(X-raydiffraction,XRD)表征
取4 mL Cur-NPs和blank-NPs冷冻干燥,将冻干的Cur-NPs和blank-NPs,以及姜黄素粉末,用XRD进行表征,观察其表面结构特征。姜黄素、Cur-NPs和blank-NPs粉末的XRD结果如图9所示,其中,姜黄素具有明显的特征峰;Cur-NPs和blank- NPs的图谱比较相似,姜黄素特征峰基本消失;由此可证明姜黄素被包裹在纳米粒中,而非吸附在纳米粒表面。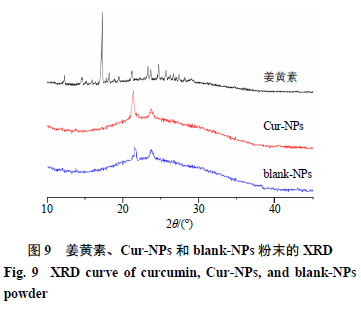
2.8 体外释放研究
将姜黄素溶液(Cur-DMSO,姜黄素用1% DMSO水溶液溶解)和Cur-NPs分别用纯化水稀释至姜黄素质量浓度为60 µg/mL,取1 mL溶液至透析袋(截留相对分子质量为14 000)中,加入200 mL 0.01 mol/L PBS(pH 7.4,含0.5%聚山梨酯80)为释放介质,置于37 ℃、100 r/min的恒温震荡箱内,在设定时间点取出5 mL释放介质,并补充相同体积的新鲜介质,测定并计算药物的累积释放率,每组平行3份。累积释放率=(CnV0+CiV)/m姜黄素
Cn为释放各时间点测得释放介质中的姜黄素的质量浓度,V为释放介质的总体积,V0为每次取样的体积,m姜黄素为透析袋中姜黄素的总质量
以PBS(pH 7.4)模拟体内循环环境,Cur-DMSO为对照,研究Cur-NPs中姜黄素的释放行为,结果如图10所示。Cur-DMSO在0~12 h内姜黄素快速
释放,在12 h累积释放率达到78%,其平均释放速率约为6.50%/h;12 h后释放趋于平缓,48 h时基本完全释放,累积释放率达到94.44%。Cur-NPs的释放过程分3个阶段:在第1阶段(0~4 h)姜黄素基本没有释放,可能由于Cur-NPs表面无姜黄素附着,姜黄素需从Cur-NPs内向外扩散才能释放,这一现象与XRD结果相符;在第2阶段(5~48 h),随着Cur-NPs向外扩散通道的形成,姜黄素平缓释放,累积释放率从0.3%增加到69.9%,平均释放速率为每小时1.62%,远低于Cur-DMSO释放速率,说明Cur-NPs具有良好的缓释能力;第3阶段(48 h后),Cur-NPs的释放速率趋于平缓,72 h时累积释放率达到80.88%。2.9 细胞毒性考察
用MTT法检测blank-NPs对U251细胞的细胞毒性,将处于对数生长期的U251细胞以每孔8×103个接种于96孔板,于37 ℃、5% CO2培养箱中孵育24 h。将blank-NPs用新鲜培养基稀释至50~ 800 µg/mL,弃去原有的培养基,每孔分别加入100 µL含blank-NPs的不同浓度培养基和新鲜培养基(对照组,细胞存活率为100%),各组平行5份,另设无细胞孔为空白组。继续培养24 h后,加入10 µL MTT(5 mg/mL)。37 ℃下继续培养4 h后,弃去旧培养基,加入150 µL的DMSO,并在酶标仪上于波长490 nm处测定吸光度(A),计算细胞存活率。细胞存活率=(A实验-A空白)/(A对照-A空白)
A实验为实验组测得的A值,A对照为对照组测得的A值,A空白为空白组测得的A值
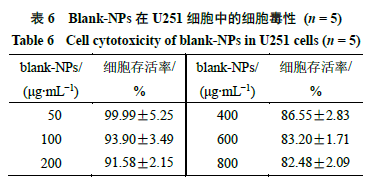
Blank-NPs的细胞毒性结果如表6所示。Blank- NPs对U251细胞的生长抑制与blank-NPs浓度呈正相关,当blank-NPs质量浓度高达800 µg/mL时U251细胞的存活率依旧保持在80%以上。表明NPs体系生物安全性良好。
2.10 细胞摄取实验
将处于对数生长期的U251细胞以每孔1×105个接种于6孔板,于37 ℃、5% CO2培养箱中孵育24 h。弃去培养基,将Cur-DMSO、Cur-NPs用新鲜培养基稀释至20 µg/mL后,加至6孔板中,每孔加入1 mL。于37 ℃、5% CO2培养箱中孵育培养3 h后,弃去含药培养基,PBS冲洗3次,荧光显微镜观察U251细胞的药物摄取情况。荧光显微镜观察U251细胞对Cur-DMSO和Cur-NPs的摄取结果如图11所示,在相同条件下培养4 h后,可以观察到Cur-NPs组细胞中的绿色荧光明显要强于Cur- DMSO组;由此可知,相比Cur-DMSO,Cur-NPs具有更强的抑制肿瘤细胞侵袭能力。
2.11 体外抗肿瘤细胞增殖实验
MTT法测定Cur-DMSO和Cur-NPs对U251细胞的抗增殖效果,细胞培养方法同“2.9”项。将Cur-DMSO、Cur-NPs用新鲜培养基稀释至药物质量浓度为2.5、5.0、10.0、20.0、40.0、60.0 µg/mL,分别加入含U251细胞的96孔板中,每孔100 µL,各浓度平行5份。另设新鲜培养基为对照组(细胞存活率为100%),无细胞孔为空白组。培养24 h后,弃去培养基,加入含10% MTT的培养基100 µL。继续培养4 h,弃去旧培养基,加入150 µL的DMSO,酶标仪测定490 nm处A值。按“2.9”项下公式计算细胞存活率,并用SPSS计算Cur- DMSO和Cur-NPs的半数抑制浓度(IC50)。在证实纳米载体良好的生物安全性基础上,进一步研究Cur-DMSO和Cur-NPs对肿瘤细胞的抗增殖能力,结果如表7所示。Cur-DMSO和Cur-NPs对U251细胞抗增殖能力均随着药物浓度的升高而上升。经计算Cur-DMSO和Cur-NPs对U251细胞的IC50分别为20.92、14.74 µg/mL,表明Cur-NPs的体外抗肿瘤活性更强。其可能原因:在相同药物质量浓度下,Cur-NPs可以更好地被摄取进入细胞并实现胞内释放,使得肿瘤细胞内具有更高的姜黄素质量浓度,因而具有更强的抗增殖能力。
3 讨论
本实验通过开环聚合和酯化反应成功合成了DPE-PCL-mPEG,采用溶剂挥发法制备了Cur-NPs,经单因素考察和Box-Behnken效应面选择最优处方,得到粒径较小,分布均匀,理化特性理想的纳米粒。体外释放和稳定性实验表明,Cur-NPs具有缓释能力,且在PBS和PBS+10% FBS中稳定性良好。体外细胞毒性实验证实,blank-NPs具有较好的生物安全性。而细胞摄取和体外抗肿瘤实验表明,针对U251细胞,相比Cur-DMSO,Cur-NPs具有更强的抗肿瘤活性和入胞能力。总之,DPE-PCL- mPEG是理想的载体材料,Cur-NPs能有效提高姜黄素生物利用度,是具有临床应用潜力的纳米给药系统;此外,该制剂的体内药学特性仍需进行更系统、深入的研究。利益冲突 所有作者均声明不存在利益冲突
参考文献(略)
来 源:洪伟勇,王金明,王海英,周雪峰,郭钫元,杨根生.载姜黄素两亲性星状聚酯纳米粒的制备、表征及体外抗肿瘤研究 [J]. 中草药, 2021, 52(8): 2237-2246 .













