
小伙伴们
大家好呀
!
听说有小伙伴因为自己的纯生信文章卡在手里一直被拒而感到苦恼?别着急,那是因为这个纯生信的“
文章公式
”
你还没有掌握!这不,馆长一听到大家有烦恼,立刻带着这篇由
暨南大学附属医院张静琳+袁永刚团队
带来的“
单细胞测序+深度学习
”的文章来和大家分享啦
!
这两个看似在纯生信文章中“平平无奇”、却又“不可或缺”的要素,是如何在大佬手里行云流水地拿下
8分+
的文章呢?馆长
也表示非常地好奇,就让我们一起来深度探索一下吧~
本文首先利用了
单细胞RNA-seq
分析技术,将原代样本与转移性样本进行了比较,并对其
肿瘤微环境
的差异进行了分析,并在
多种算法
帮助下对关键基因进行了分析,以完成对UVM患者
深度学习
预后模型的建立。那么,本文究竟是如何合理将多种算法与多数据库进行结合,成功以纯生信拿下这篇高分文章的呢?它与其它的纯生信文章相比,究竟有哪些“过人之处”呢?就让馆长来带大家好好分析一下吧~
1.
近年来,
单细胞RNA-seq
分析几乎快要成为了纯生信领域的“大王”,而它主要的一大优点便是可以从
肿瘤微环境
的角度对不同的样本进行分析,本文正是利用了这一特点,对不同样本间巨噬细胞的富集情况进行了对比与分析,从而加深读者对于UVM肿瘤微环境的理解;
2.
基于
多种算法
对
多数据库
进行分析,作者成功提取出UVM中的关键基因,并将
机器学习与深度学习
进行结合,开发了一款可以显著区分原发性患者与转移性患者的预后模型,它不仅可以用于分类,还可以用于预测与生存分析,这为UVM患者的个性化治疗提供了极大的帮助;
3.
除了上述的热点之外,还有一样统计方法引起了
馆长
的关注——那便是
HdWGCNA
这一分析方法,相较于传统的WGCNA方法,HdWGCNA更适用于处理单细胞测序一类的
高维度数据
,同时能为数据后续的处理提供更精准的共表达网络,在
多组学数据
日益丰富的今天,HdWGCNA应用的前景也会越来越广阔。
方法太难了学不会?数据太多太乱,不知道该用哪种方法分析?别担心,
馆长
来帮你处理!不要浪费了自己手中的大好数据~快快扫描下方二维码,让
馆长
来解决你的问题,下一个高分文章之王就是你~
定制生信分析
云服务器租赁
加好友
备注“99”
领取试用

题目:单细胞 hdWGCNA 揭示葡萄膜黑色素瘤的转移性保护性巨噬细胞与其深度学习预测模型的开发
杂志:Journal of Translational Medicine
影响因子:IF=8.44
发表时间:2024年8月
公众号回复“123”领取原文PDF,文献编号:
20240902
研究背景
作为目前最常见的原发性成人眼肿瘤,葡萄膜黑色素瘤(UVM)的发病率通常受多种因素,如年龄、种族和纬度影响。美国数据显示,约50%的原发性UVM患者会发展为转移性疾病,而局部治疗方法目前来看仅对原发性UVM有效,对转移性UVM尚且无法有效治疗。研究发现,巨噬细胞在肿瘤免疫中扮演重要角色,不同亚群的巨噬细胞对肿瘤有不同影响。本研究使用单细胞 RNA-seq 分析了原发性UVM与转移性UVM之间巨噬细胞富集的区别,并基于关键基因建立了全新的UVM患者预后模型,为UVM的治疗和预后提供了新的策略。
数据
来源
作者收集了来自多个数据集的公共单细胞转录组学与患者数据,包括但不限于GEO、UCSC Xena、TCGA 数据库中的数据,为该机器模型的建立提供了丰富的数据来源。
|
数据集/队列
|
数据库
|
数据类型
|
详细信息
|
|
GSE22138
|
GEO
|
UVM患者数据集
|
包含28 个原始样本和 35 个转移样本
|
|
GSE58294
|
GEO
|
UVM患者数据集
|
包含 18 个原始样本和 11 个转移样本
|
|
-
|
UCSC Xena
|
GDC TCGA
UVM数据
|
-
|
|
GSE139829
|
GEO
|
UVM 单细胞数据集
|
9 个原代非转移样本和远端转移后组织样本
|
|
-
|
TCGA 数据
|
UVM患者数据集
|
80例UVM患者
|
研究思路
这篇文章首先对UVM样品进行了单细胞RNA-seq分析,发现原代肿瘤样本与转移性UVM间肿瘤微环境存在一定的差异,在原代肿瘤样本中发现了巨噬细胞的富集。接下来,作者使用HdWGCNA方法对转移性UVM样本中的巨噬细胞关键基因进行了鉴定与分析,并利用多种算法对这些MPMφ 基因的功能、交互作用网络与样本聚类进行分析,根据分析得到的结果,作者进行了进一步的验证,并将机器学习与深度学习进行结合,得到了能够对UVM患者进行分类与风险评估的预后模型,并用公开数据库对其功能进行了验证,证明了该预后模型在UVM患者预后方面的可靠能力,为UVM未来的个性化治疗提供了一定的帮助。
研究结果
1.原发性与转移性UVM样本单细胞测序分类
通过对原发性和转移性UVM样本进行
scRNA-Seq分析
,作者识别了28个不同的细胞集群,最终确定了 8 个主要的细胞簇以及它们的标记基因(图1A,B)。根据
肿瘤微环境分析
发现,原发性UVM患者的
单核细胞与巨噬细胞细胞比例
比转移样本更高(图1C,D)。进一步对两组数据的聚类分析与UMAP图进行比较,可以发现原发组和转移组在特定子簇中的细胞数量存在着
显著的差异
(图1E,F)。
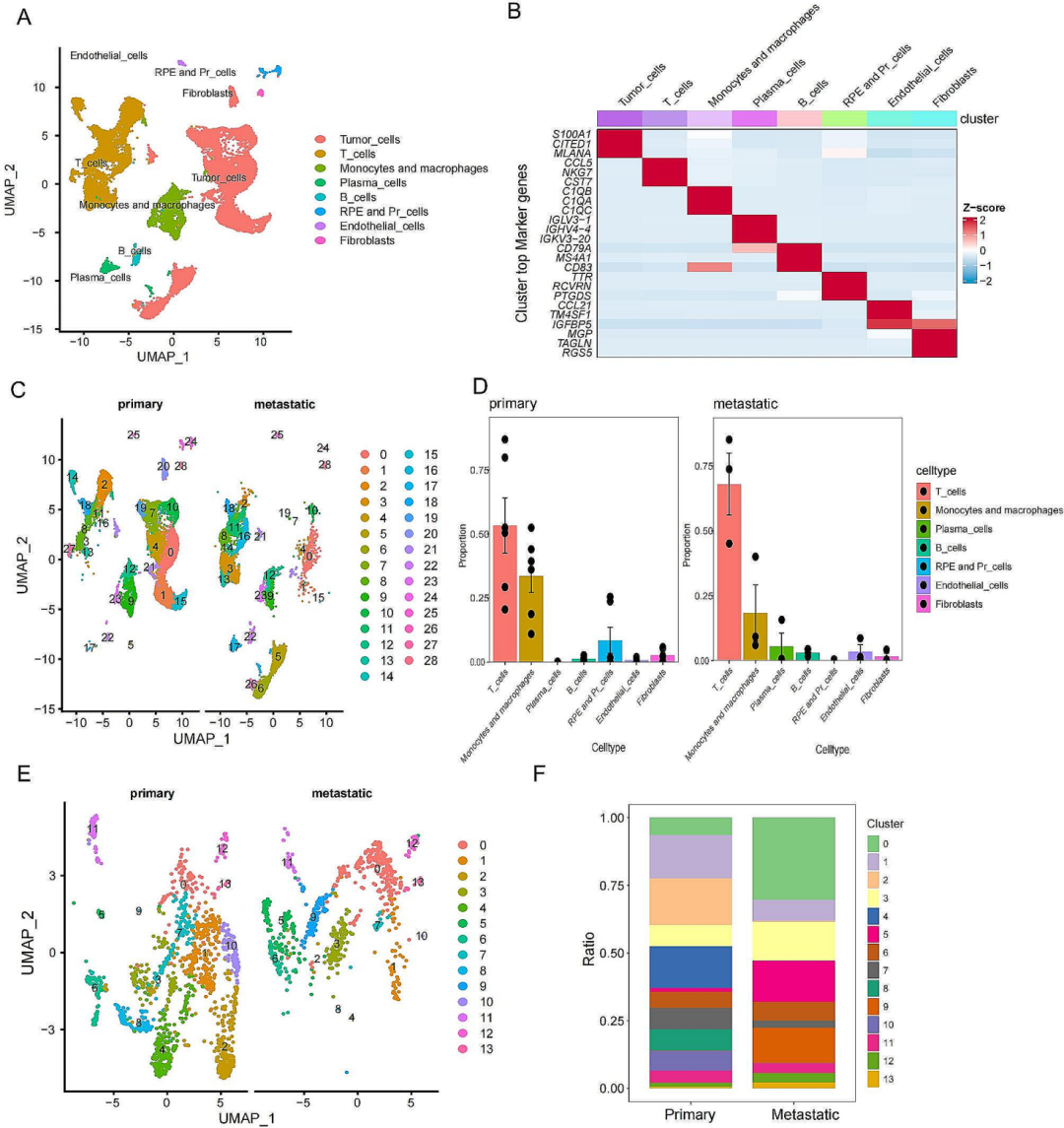
图1
UVM样品中巨噬细胞的单细胞RNA-seq分析
2.MPMφ的鉴定与表征
根据已有的文献,作者将单核细胞与巨噬细胞分为6类,并对其进行了
伪时间分析
,发现这些细胞在
不同的分化阶段
表现出了
不同的基因表达特征
(图2A-D)。为了方便区分,作者将感兴趣的细胞簇命名为“primary_Mac”,并对
细胞间通信
进行了分析,结果发现primary_Mac在CXCL信号通路中作为发送者和影响者均发挥了较为重要的作用(图3A-F)。
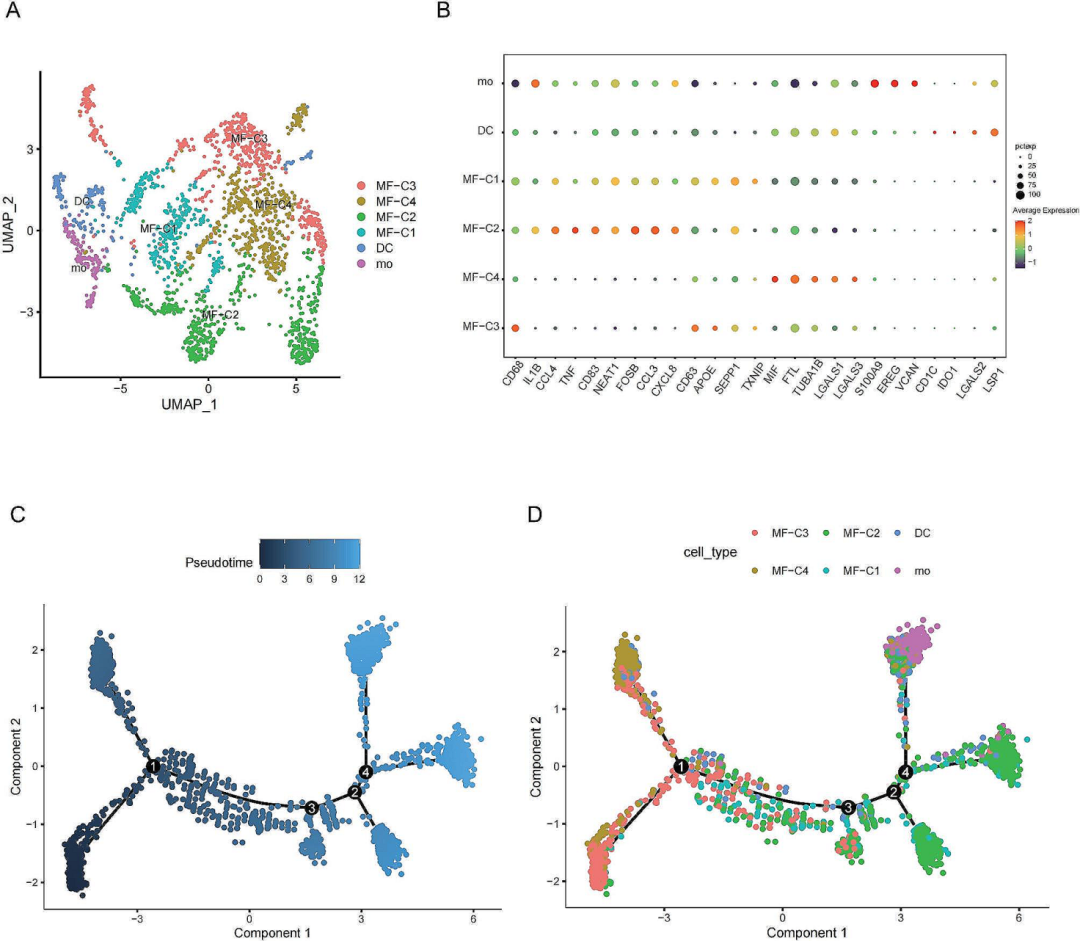
图2
巨噬细胞亚聚类和Monocle结果
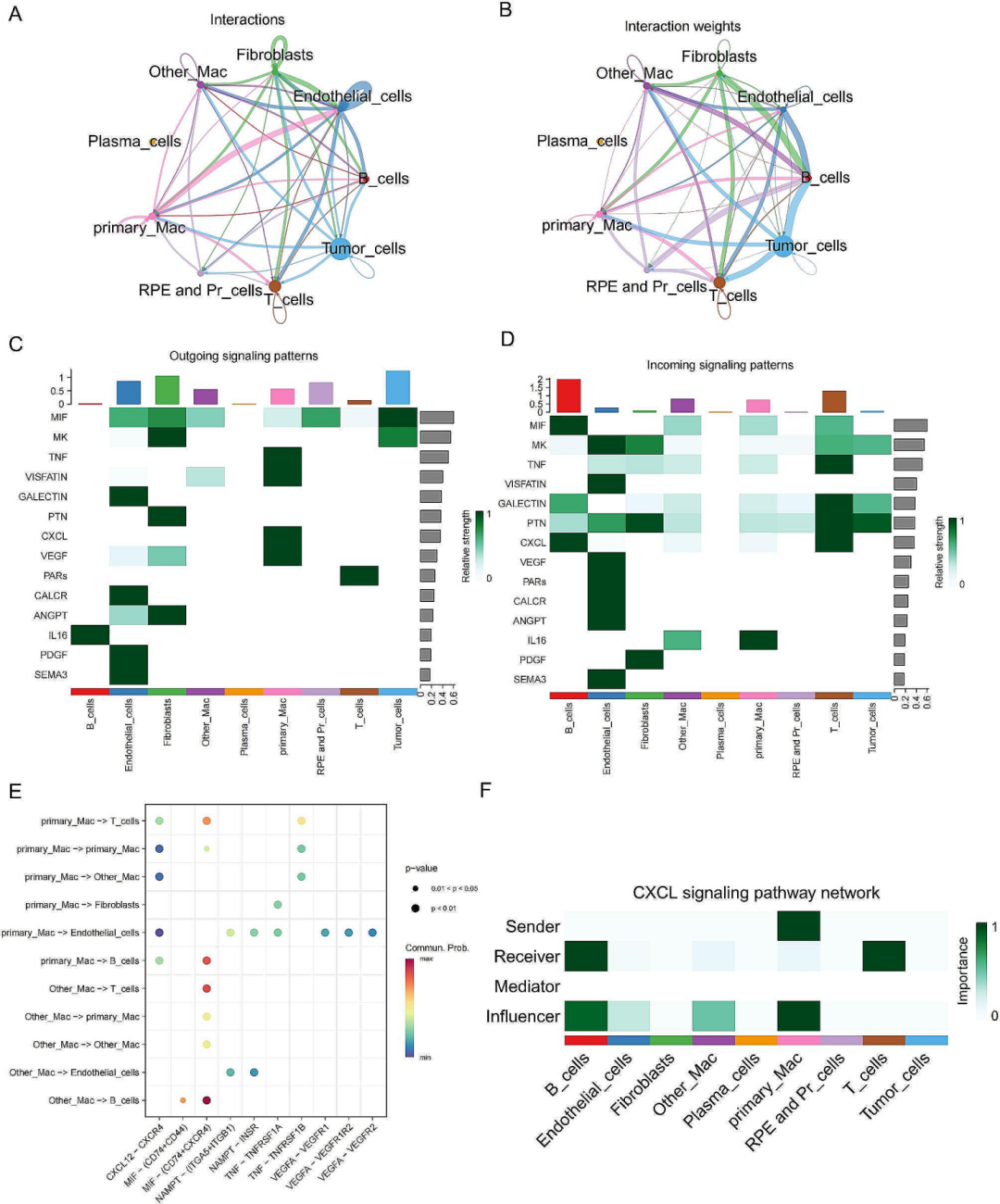
图3所有细胞类型的全面CellChat分析
3.HdWGCNA进行MPMφ相关关键基因与功能通路分析鉴定
作者使用了
高维加权基因共表达网络分析(HdWGCNA)
对原发性巨噬细胞的关键分子特征进行了鉴定,确定了四个基因模块与它们之间的相关性(图4C),其中蓝绿色模块与棕色模块在细胞簇2、4、8、10中高度激活(图4D,E)。作者进一步计算了模块之间的连接性,并确定了
这两种模块与原发性巨噬细胞簇密切相关
(图4F)。
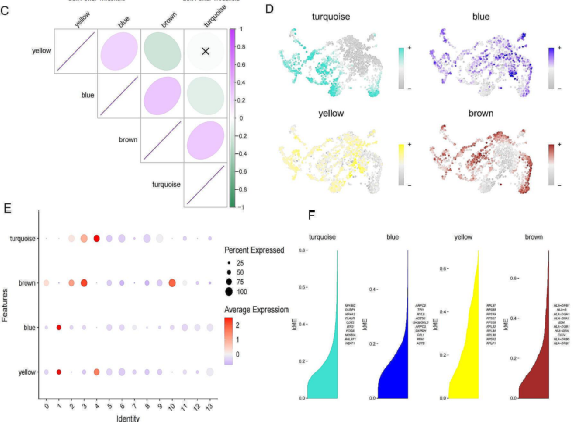
图
4
巨噬细胞的
Hd
WGCNA分析
4.聚类分析
从前一步确定的两个模块中,作者各选择了25个MPMφ关键基因,并在UVM患者数据集中进行
NMF聚类分析
,将患者分为两个不同的亚型(图5B)
。
GSE分析显示,这两种基因在2型患者中的表达
显著高于
1型(图5C)。进一步分析显示,
大多数基因
均在2型患者中
高表达
,而在1型患者中处于
低表达
的状态(图5D)。
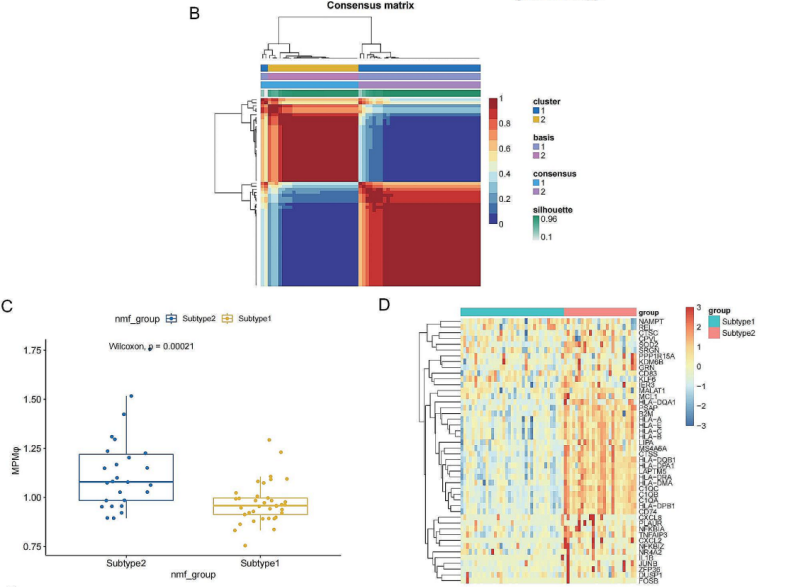
图
5 UVM中MPMφ细胞亚型的鉴定









