
RTKs是最大的一类酶联受体,已发现有50多种不同的RTKs家族成员,其中主要包括FGFRs (Fibroblast Growth Factor Receptor,FGFR)、表皮生长因子受体(epidermal growth factor receptors,EGFRs)、血小板衍生生长因子受体(platelet-derived growth factor receptors,PDGFRs)、血管内皮生长因子受体(vascular endothelial growth factor receptors,VEGFRs)和肝细胞生长因子受体(hepatocyte growth factor receptors,HGFRs)。
成纤维细胞生长因子受体家族(FGFR)属于受体激酶家族,它包括由四种密切相关的基因所编码的四种受体亚型(FGFR-1,2,3 和4)及一些异构分子,它们通过与18种不同的成纤维细胞生长因子(FGF)形成三元复合物,进而引发一系列的信号传导途径,参与调节生物体内的生理过程,如:胚胎发育(embryonic and fetal development),伤口愈合(wound healing)和血管生成(angiogenesis)等。FGFR下游涉及多条不同功能的信号通路,如下图所示。
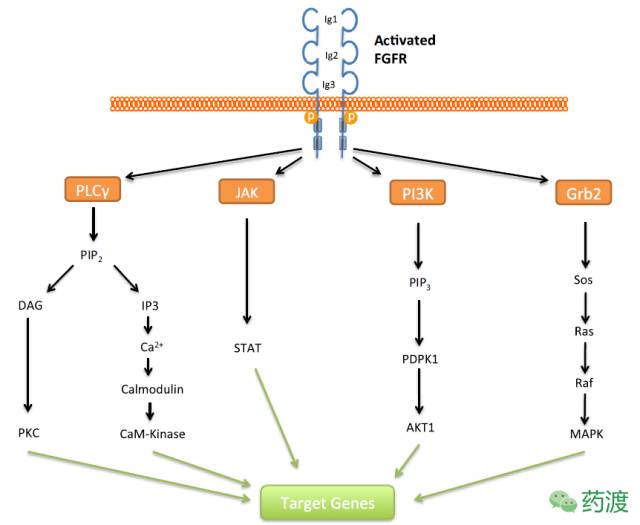
Signalling pathways that may be activated upon FGF ligand binding and receptor dimerization
与表皮生长因子(EGFR)类似,FGFR信号通路的异常也已成为多种肿瘤重要的成因,具体机制如下图所示。在一项针对210种人类肿瘤细胞518种蛋白激酶基因编码区的研究中,科研人员发现FGFR信号通路包含了最多的非同义突变。近期一项对4853例实体瘤案例的分析表明,FGFR的功能异常发生于7.1%的肿瘤样本中,其中基因扩增、基因突变以及染色体重排分别占比为66%、26%及8%;FGFR1、FGFR2、FGFR3及FGFR4在发病患者中占比为3.5%、1.5%、2.0%及0.5%。另外,FGFR信号通路的激活也会导致一些肿瘤细胞产生对靶向治疗的抗性。因此,FGFR也是靶向药物研发的一个热点领域。本文将对目前已上市的及正处于不同研发阶段的靶向于FGFR信号通路的抑制剂类药物做一简要盘点。
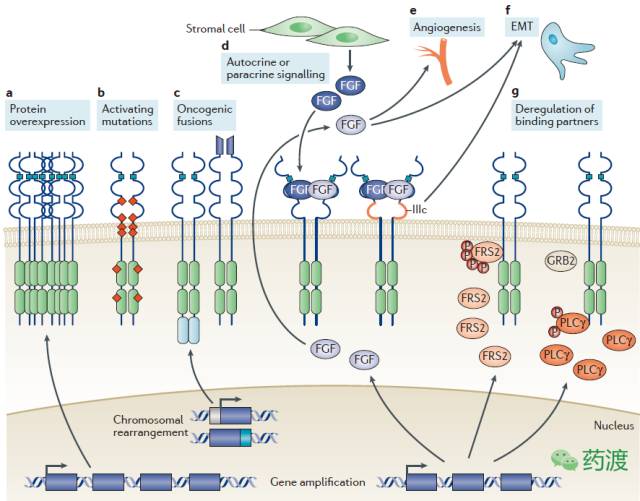
Mechanisms of oncogenic fibroblast growth factor receptor signalling
图注:
a,FGFR基因扩增并表现为对应蛋白分子的过表达,从而导致FGF受体的累积及下游信号的过分激活。b,激活突变,通常导致配体缺失状态下受体的二聚化增加,或者激酶结构域的不受控持续激活状态。
c,染色体转位,从而导致FGFR分子与其它蛋白融合或处于不同蛋白的启动子区而引发受体异常激活。d,肿瘤细胞自分泌及旁分泌导致的FGFR过量激活。
e/f,肿瘤及相关细胞分泌的FGF诱导产生新生血管及EMT过程。
g,FGFR结合蛋白的调控异常导致下游信号通路的异常激活。
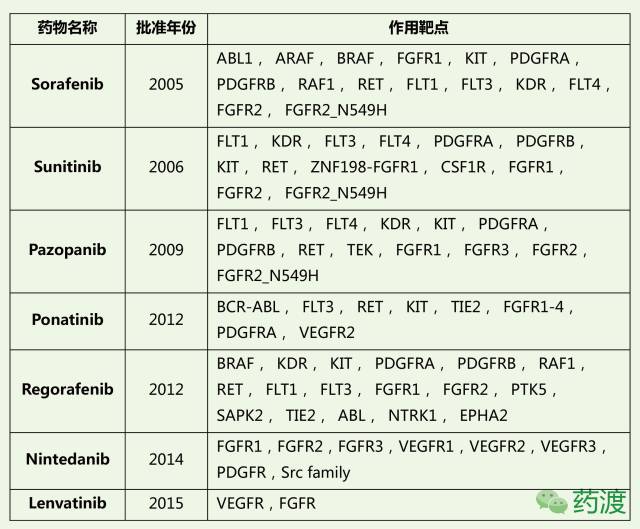
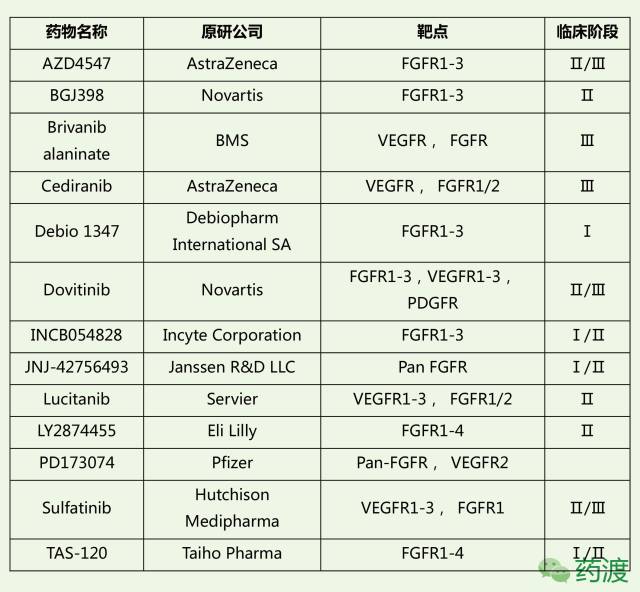
迄今为止,美国FDA总共批准了7类FGFR抑制剂类药物,具体信息如下表所列。
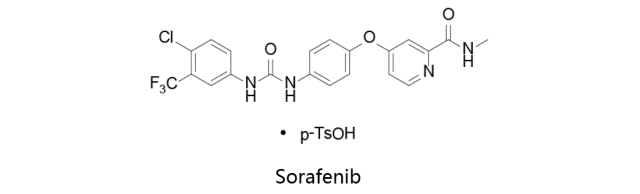
Sorafenib:
甲苯磺酸索拉非尼由拜耳和Onyx联合研发,研发代码BAY 43-9006,于2005年12月20日首次获得美国FDA批准上市,此后于2006年7月19日获得EMA批准上市,于2008年1月25日获得日本PMDA批准上市,并由拜耳上市销售,商品名确定为Nexavar。索菲拉尼是一种激酶抑制剂,能够同时抑制多种存在于肿瘤细胞并参与肿瘤细胞信号转导、血管生成和细胞凋亡的胞内激酶(c-CRAF,BRAF和突变型BRAF)和细胞表面激酶(KIT,FLT-3,RET,RET/PTC,VEGFR-1,VEGFR-2,VEGFR-3及PDGFR-β)。该药适用于治疗不能切除的肝细胞癌、晚期肾细胞癌以及局部复发或转移性、渐进性、分化型并且难以用放射性碘治疗的甲状腺癌等。Sorafenib是Raf-1抑制剂,对Raf-1,B-Raf和VEGFR2的IC50分别为6 nM,22 nM和90 nM。
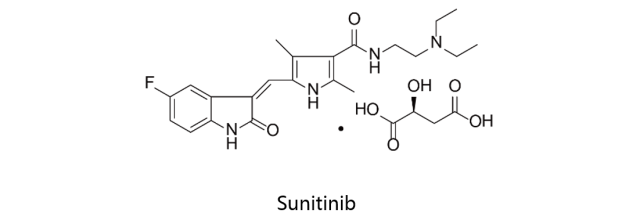
Sunitinib:
苹果酸舒尼替尼由Pfizer研发并生产,首先于2006年1月26日获得美国FDA批准上市,之后分别于2006年7月19日及2008年4月16日获得EMA及PMDA批准,由辉瑞上市销售,商品名确定为Sutent。舒尼替尼是多靶点受体酪氨酸激酶(RTKs)小分子抑制剂,具有抑制肿瘤血管生成和抗肿瘤细胞生长和转移的多重作用。该药物用于治疗胃肠道间质瘤(GIST),晚期肾细胞癌(RCC)和胰腺神经内分泌肿瘤(pNET)。Sunitinib对PDGFRβ及VEGFR2的IC50值分别为2nM及80nM。
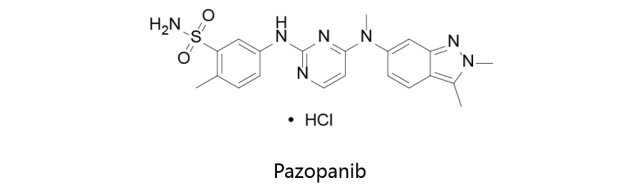
Pazopanib:
盐酸帕唑帕尼由GSK开发,首先于2009年10月9日获得美国FDA批准上市,之后分别于2010年及2012年获得EMA及PMDA批准上市,由GSK在美国、英国和日本市场销售,商品名定为Votrient。该药物是一种多靶点的受体酪氨酸激酶抑制剂,对VEGFR1,VEGFR2,VEGFR3,PDGFR,FGFR,c-Kit和c-Fms的IC50分别为10nM,30nM,47nM,84nM,74nM,140nM和146nM。在体外,Pazopanib抑制血管内皮细胞生长因子VEGFR-2、细胞因子受体Kit和血小板衍生生长因子PDGFR-β的配体诱导的自磷酸化;在体内可抑制小鼠肺中VEGF诱导的VEGFR-2的磷酸化、小鼠体内的血管生成及异种移植的人类肿瘤细胞的生长。该药物适用于有既往化疗经历患者的晚期肾细胞癌和晚期软组织瘤的治疗。
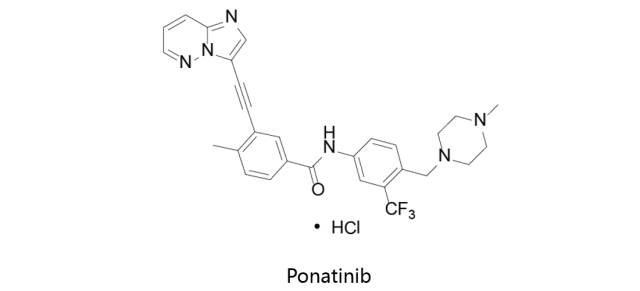
Ponatinib:
由Ariad Pharm研发,首先于2012年12月14日获得美国FDA批准上市,之后分别于2013年及2016年获得EMA及PDMA批准,由Ariad在美国和欧洲上市销售,商品名为Iclusig。Ponatinib (AP24534)是新型的多靶点抑制剂,对Abl,PDGFRα,VEGFR2,FGFR1和Src的IC50分别为0.37nM,1.1nM,1.5nM,2.2nM和5.4nM。该药物用于治疗对既往酪氨酸激酶抑制剂治疗耐药或不耐受的慢性粒细胞白血病(CML),或对既往酪氨酸激酶抑制剂治疗耐药或不耐受的费城染色体阳性急性淋巴母细胞白血病(Ph+ALL)。
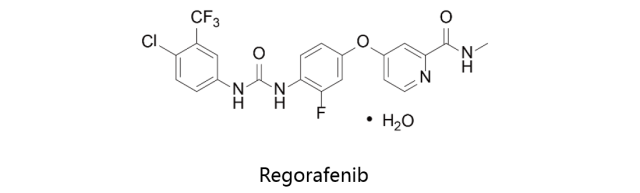
R
egorafenib:
由拜耳研发,首先于2012年9月27日获得美国FDA批准上市,之后分
别于2013年3月25日及8月26日获得PMDA及EMA批准上市,商品名定为Stivarga。Regorafenib (瑞格非尼;BAY 73-4506)是多靶点抑制剂,对VEGFR1,VEGFR2,VEGFR3,PDGFRβ,Kit,RET和Raf-1的IC50分别为13nM,4.2nM,46nM,22nM,7nM,1.5nM和2.5nM。该药物用于治疗转移性结肠直肠癌(CRC)和局部晚期无法手术切除或转移性胃肠道间质瘤(GIST)。
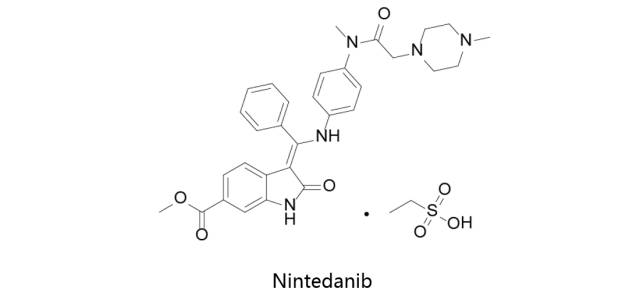
Nintedanib:
乙磺酸尼达尼布由Boehringer Ingelheim研发,于2014年10月15日获得美国FDA批准上市,并于2014年11月21日及2015年7月3日获得EMA及PMDA批准上市,商品名为Ofev(美国)/Vargatef(欧洲)。该药物是一种多重酪氨酸激酶抑制剂,Nintedanib (BIBF1120)是三重血管激酶抑制剂,对VEGFR1/2/3,FGFR1/2/3和PDGFRα/β的IC50分别为34nM / 13nM / 13nM,69nM / 37nM / 108nM和59nM / 65nM。通过抑制与特发性肺纤维化(IPF)的发病机制相关的生长因子受体而起作用。先后获批治疗特发性肺纤维化和非小细胞肺癌。
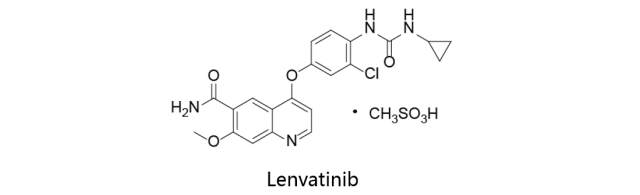
Lenvatinib:
由Eisai研发,与2015年2月13日获得美国FDA批准上市,同年3月26日及5月28日分别获得PMDA及EMA批准,商品名定为Levima。该药物是一种口服多受体酪氨酸激酶抑制剂,具有独特的结合模式,可选择性抑制血管内皮生长因子VEGF受体激酶活性,此外还可抑制参与肿瘤细胞增殖的其它促血管生成和致癌信号通路相关的酪氨酸激酶。Lenvatinib (E7080)是多靶点抑制剂,对VEGFR2 (KDR)和VEGFR3 (Flt-4)的IC50分别为4nM和5.2nM,对VEGFR1 / Flt-1,FGFR1和PDGFRα/β的抑制性较弱。该药适用于复发或进展性及放射性碘难以治疗的分化型甲状腺癌的治疗。
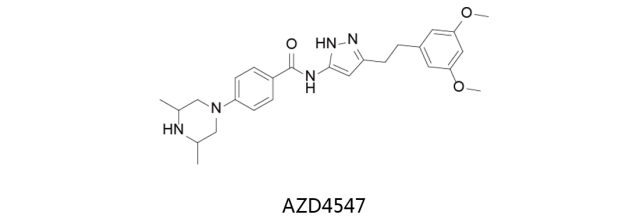
AZD4547:
新型的口服FGFR选择性抑制剂,对FGFR1、FGFR2和FGFR3体外测定的IC50值分别为0.2、2.5及1.8nmol/L。AZD4547对VEGFR也有抑制活性,但活力比对FGFR1低约100倍。该药物由阿斯利康研发,目前处于临床二期和三期研究中,用于治疗实体瘤及非小细胞肺癌。
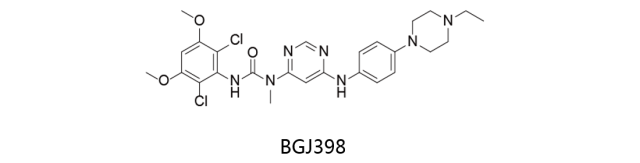
BGJ398:
又名Infigratinib,诺华研发。一类广谱的FGFR抑制剂,对FGFR1抑制活力最强,IC50值小于10nmol/L;对VEGFR也表现出抑制活性,但活力比对FGFR1-3低约70-100倍。目前处于临床二期阶段,用于治疗黑色素瘤和复发性胶质母细胞瘤。另一项对于治疗FGFR突变型癌症(包括FGFR突变型晚期实体瘤,FGFR基因改变型晚期或转移性胆管癌,FGFR基因改变的恶性血液性肿瘤)的研究也处于二期临床阶段。最常见的不良事件包括胃肠道事件和疲劳。
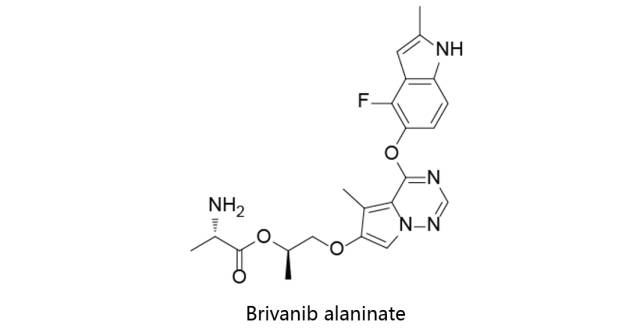
Brivanib alaninate:
又名BMS-582664,由百时美施贵宝原研,目前进入三期临床用于治疗肝癌。2011年,EMA因治疗肝细胞癌授予该药孤儿药。2015年再鼎医药获得该药物在中国地区的研发权利,并于2016年7月向CFDA提交了临床申请(1类化药)。
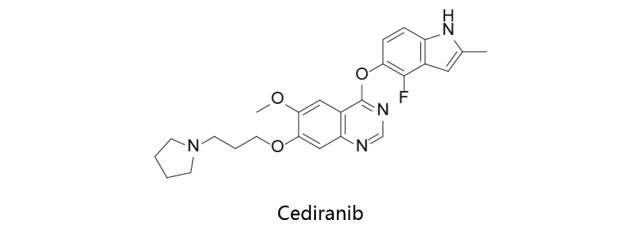
Cediranib:
阿斯利康原研,又名AZD2171,是一种泛血管内皮生长因子(pan-VEGF)受体酪氨酸激酶抑制剂。主要抑制VEGFR-1、VEGFR-2、VEGFR-3和PDGFR。西地尼布可抑制3种血管内皮生长因子(VEGF)受体,作用于血管和淋巴管,发挥抗血管生成作用,抑制肿瘤的生长和扩散。
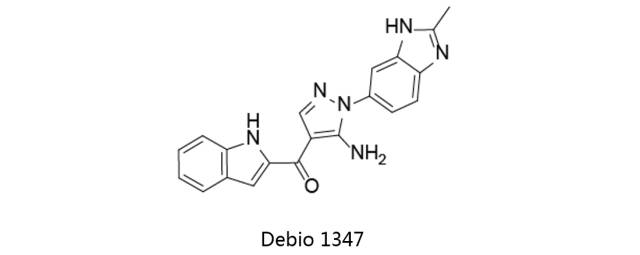
Debio1347:
又名CH-5183284,最初由罗氏原研,后授权Debiopharm全球开发权。目前该药处于临床一期阶段,用于治疗晚期实体肿瘤。Debio1347为口服FGFR选择性抑制剂,对FGFR1、FGFR2、FGFR3及FGFR4的IC50值分别为9.3、7.6、22及290nM。
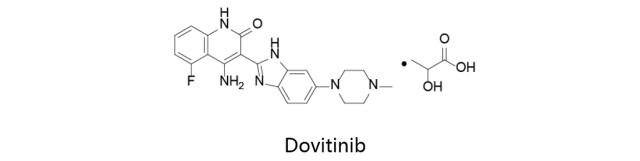
Dovitinib:
又名TKI258/CHIR258,由诺华公司原研,目前处于三期临床阶段,用于治疗肾细胞癌和实体瘤,但该项研究目前已被终止。目前针对转移性前列腺癌,非小细胞肺癌,乳腺癌,结直肠癌,胃肠道间质瘤,肝癌,甲状腺癌及胰腺癌等适应症的研究正处于临床二期阶段。该化合物因治疗颌下腺腺样囊性癌与2013年获得美国FDA授予的孤儿药资格。Dovitinib为多靶点RTK抑制剂,对FLT3/c-Kit抑制的IC50值为1/2nM,对FGFR1/3的IC50值在8-13nM左右。
INCB054828:
Incyte原研药物,目前处于临床一期和二期阶段,用于治疗实体肿瘤和多发性骨髓瘤。该化合物为强效FGFR1/2/3抑制剂,对携带FGFR功能异常的肿瘤细胞具有选择性药理活性。
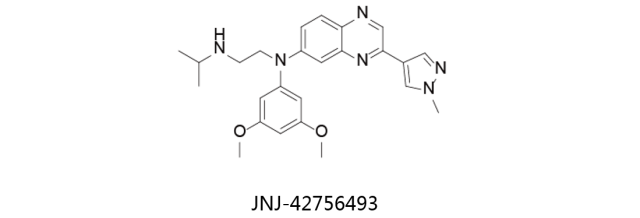
JNJ-42756493:
又名Erdafitinib,由杨森研发,处于临床二期用于治疗膀胱癌和肝癌。对于该化合物的另一项研究处于临床一期,用于治疗晚期难治性实体瘤与血液癌症。Erdafitinib (JNJ-42756493)是有效的、具有选择性和口服生物活性的泛成纤维细胞生长因子受体FGFR抑制剂,具有潜在的抗肿瘤活性。对FGFR家族所有成员(FGFR1-4)的IC50都在低浓度摩尔范围内,而对VEGFR的活性影响甚微。
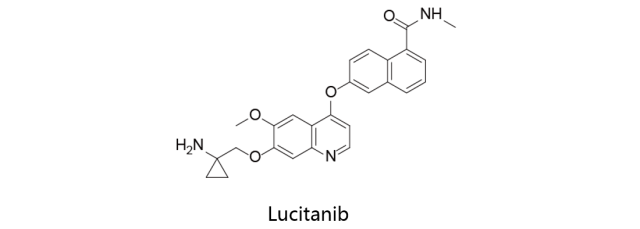
Lucitanib:
最初由爱德程研发,2008年授权给EOS Pharmaceuticals,2012年后者授权给施维雅(除美国,日本和中国外)。2013年,施维雅与中科院上海药物所大成在中国研发该化合物的协议。Lucitanib目前处于临床二期,用于治疗FGF-异常转移性乳腺癌,晚期或转移性非小细胞肺癌,小细胞肺癌或其他实体瘤。该药物为蛋白激酶抑制剂,可一直VEGFR1/2/3,FGFR1/2及PDGFR-α/β的活力。
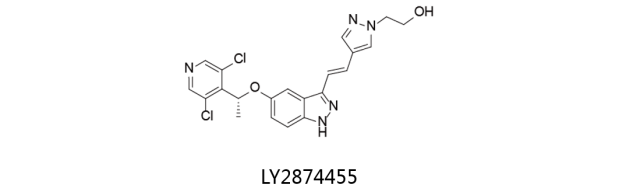
LY2874455:










