Mfuzz是较有知名度的聚类R包,它采用的模糊均值算法是普通K-means算法的改进。普通K-means算法对于数据的划分是硬性的,让它聚成20个类,结果就会出现20个类。Mfuzz则是一种柔性的模糊划分,让它聚成20个类,结果可能是16个类,也可能是25个类。K-means的硬聚类把每个待辨识的对象严格地划分到某类中,具有非此即彼的性质。Mfuzz的模糊聚类建立了样本对类别的不确定性描述,会根据数据本身的特性来调节聚类结果,从而更有优势。脚本如下:
## 安装 ##
source("https://bioconductor.org/biocLite.R")
biocLite("Mfuzz")
## 输入数据 ##
library("Mfuzz")
rawdata读入csv文件
colnames(rawdata)=c("geneID","IR1","IR2","IR4","IR5","IR6")# 文件有6列,添加列名
gennames第一列是基因名字
rn
rownames(rawdata)=rn # 行名
rawdata
rawdata
eset
## 过滤数据 ##
mydata.r
mydata.f
tmp
mydata.s
# 根据我的经验,这一步可能产生新的NA,导致下面的步骤报错,解决方法之一就是重新# 运行上面的过滤步骤,把筛选参数变得更严格,尽量去掉噪音基因。
## 筛选聚类参数 ##
m
tmp
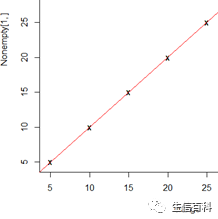
# 计算c值,即该数据应该聚多少类
# 如下图,X与红线分离的x轴,就是c值,(因为输入文件没有规律,下图未出现分离)
## 软聚类 ##
cl
mfuzz.plot2(mydata.s, cl = cl, mfrow =c(4,3), time.labels = c("IR1", "IR2", "IR4", "IR5", "IR6"), xlab="Period", ylab="Exp",colo="fancy", ax.col="red", col.sub = "blue", x11 = FALSE, col = "blue")
#聚类的结果太多,可能报错:画板太大,无法展示图片。
## 聚类细节 ##
cl[[4]][ ,1] # cluster 1的所有基因列表及其与该类的关系值
cl[[2]] # 每个cluster的基因数量
plot(cl[[1]][8,],type="l",ylab="Expression")# cluster 8的聚类中心及走势
# 下面的步骤是单独画某一cluster,(single=1:cluster 1)
mat
l
mfuzz.plot2(mydata.s,cl=cl,mfrow=NA,colo="fancy",ax.col=1,col="blue",cex.main=2,single=1,x11=FALSE,time.labels=c("IR1","IR2","IR4","IR5","IR6"))
mfuzzColorBar(col="fancy",main="Membership",cex.main=1)
#
Mfuzz需要两个参数:聚类数目c和参数m。一般来讲C要远远小于聚类样本的总个数,同时要保证C>1。对于m,它是一个控制算法的柔性的参数,如果m过大,则聚类效果会很次,而如果m过小则算法会接近硬聚类算法。参数m又称为平滑因子,控制着模式在模糊类间的分享程度,因此要实现模糊聚类就必须选定一个合适的 m值。