前言
近些年,CRISPR-Cas系统频频登上“CNS”,其强大的基因编辑能力迅速让其从基础研究转化到临床试验。随着越来越多CRISPR-Cas家族成员的发现以及基础研究的进展,CRISPR的应用范围也越来越广。今天,小编就详细介绍下Class 2家族CRISPR的作用机制,以及它们在基因编辑,靶点发现,核酸 检测等多方面的进展。
CRISPR系统简介与分类
为了应对外来病毒和质粒的入侵,细菌和古生菌进化出了一套获得性免疫系统CRISPR-Cas。CRISPR-Cas系统主要包括由Cas基因组成的操纵子和CRISPR阵列,其中CRISPR由外来基因组靶标序列(spacers)和相同的重复序列(direct repeat)所构成。CRISPR-Cas系统发挥作用主要包括Adaptation,crRNA maturation以及Interference三个过程:首先在Cas1,Cas2等蛋白的作用下,将外来DNA序列组装到CRISPR阵列中,接着在系统外的RNase III或者系统自身效应蛋白的作用下,完成pre-crRNA的切割,最后效应蛋白在crRNA的引导下靶向目的DNA/RNA,从而发挥干扰作用。与Class 1 CRIPSR需要多个蛋白构成效应复合物不同,Class 2 CRISPR只需要单个效应蛋白。根据效应蛋白的不同,Class 2又可以进一步分为三个不同类型,分别是以Cas9为代表的Type II,以Cpf1为代表的的Type V和以C2c2为代表的的Type VI。
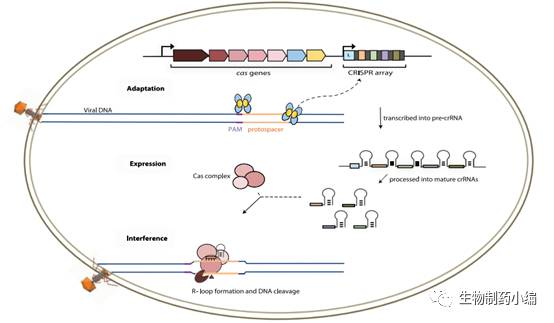
▲
CRIPSR-Cas
系统示意图
Class2 CRIPSR简介:Cas9,Cpf1与C2c2
CRISPR-Cas9是目前为止研究最深,应用最广的CRIPSR系统,它与后发现的Type V家族Cpf1都能够靶向目的dsDNA,但是两者相比有以下明显不同:1. Cpf1只需要一个crRNA完成目的序列的靶向,而Cas9中需要crRNA和tracrRNA两个分子来完成;2. Cpf1识别3’富含T的PAM序列,而Cas9识别5’富含G的PAM序列;3. 在Cas9中有HNH和RuvC两个活性结构域分别切割目标DNA的互补链和非互补链,而在Cpf1中一个全新的活性结构域Nuc,和RuvC分别切割目标DNA的互补链和非互补链;4. Cas9在PAM临近的位置切割DNA产生平末端,而Cpf1在PAM远端切割DNA产生突出的末端。而Type VI家族C2c2是一个RNA-guided的RNase,它含有两个HEPN结构域。在识别了目的RNA后,C2c2就变成了一个非特异的RNase,因此导致细胞毒性与程序性死亡。
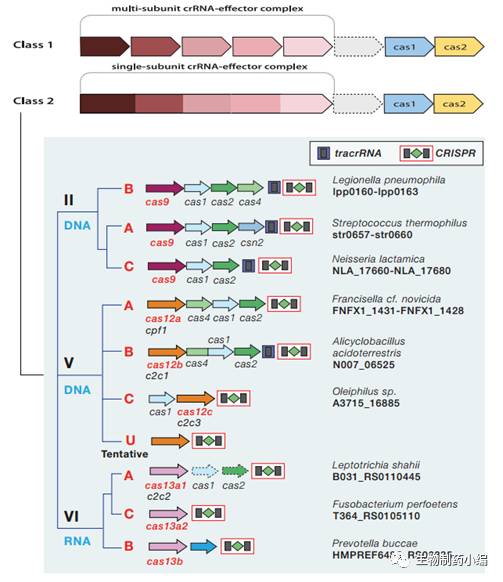
▲
Class II CRISPR
分类
随着Cas9, Cpf1, C2c2的结构被解析,人们对于这些蛋白的机制和应用理解更深入。CRISPR技术引领了一波生物技术革新,由于其精确靶向性和易操作性,Cas9和Cpf1被广泛应用于基因编辑于基因治疗,而C2c2也能够用于基因沉默与核酸检测。这些技术将在下文向大家介绍。
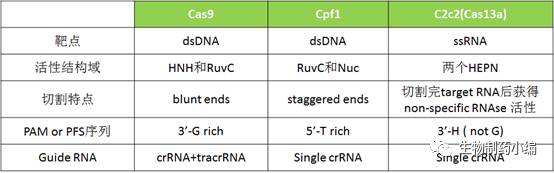
▲
Cas9
,
Cpf1
与
C2c2
的特点
CRISPR
与基因编辑
虽然大部分
Class 2 CRISPR
蛋白在体外都具有切割活性,然而只有少数被证明能够用于哺乳动物细胞的编辑
。此外,由
PAM
带来的序列限制性,
DNA
特异性带来的脱靶现象等,也是基因编辑中亟待解决的问题。

▲
可用于哺乳
动物细胞基因编辑的
CRISPR
酶与其
PAM
序列
PAM
序列特异性的改造
。
以最常用的
SpCas9
为例,其
PAM
序列
NGG
在人类基因组上平均每
8-12bp
就会出现一次,这个频率基本能满足经典的
HDR
或者
NHEJ
为基础的基因编辑。然而对于一些需要单个核苷酸分辨率的情况,或者如
SaCas9
中,
PAM
序列为
NNGRRT
,此时的
PAM
特异性就会成为一个重要的限制。为此,如何减少
PAM
的限制,扩展
Cas9
的靶向范围是非常具有意义的。
现在已有一些工作,通过突变Cas9蛋白的一两个氨基酸,从而改变其PAM的特异性。野生的FnCas9的PAM序列为NGG,而E1369R/E1449H/R1556A三突变RHAFnCas9则可将PAM序列放宽至YG。
通过细菌选择系统筛选得到的突变体VQRSpCsa9 (D1135V/R1335Q/T1337R) 可识别PAM为NGA和NGCG,EQR SpCas9(D1135E/R1335Q/T1337R)可以识别NGAG,VRERSpCas9 (D1135V/G1218R/R1335E/T1337R)可识别NGCG。
而在SaCas9中,突变体KKH(E782K/N968K/R1015H)能够将原PAM序列NNGRRT放宽到NNNRRT,而不增加SaCas9的脱靶频率。
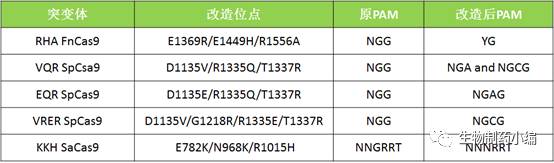
▲
PAM
序列特异性的改造
降低脱靶效应
。
作为一种基因编辑工具,脱靶效应会产生大量副产物从而影响实验效率,因此如何降低脱靶效应也是值得不断探索的技术难题。有研究显示,
Cpf1酶的特异性比Cas9高
,因此使用CRISPR-Cpf1系统进行基因编辑可以降低脱靶效应。对于SpCas9及其他Cas9,
减少sgRNA与目的DNA的互补序列至20个bp以下
,可以显著提高Cas9的特异性。减少Cas9蛋白在细胞中的存在时间也能够提高特异性,比如直接递送Cas9:sgRNA核苷酸蛋白复合物(RNPs)相比于质粒转染,产生的脱靶效应更少。此外使用改造的Cas9也能够特异性。比如
使用两个单活性结构域失活的nickase Cas9(Cas9n)
,分别靶向目的基因改造区域的两侧,这种方法能在保持on-target活性的基础上降低几个数量级的脱靶效应。或者
使用融合了非特异性限制内切酶FokI的失活Cas9(dCas9)
,FokI需要二聚才能发挥功能,当两个dCas9分别靶向目的基因改造区域的两侧,FokI才能在特定位点发生二聚从而切割DNA。此外,
使用内含肽失活的Cas9系统(intein-inactivatedCas9)
,
或者small-molecule-dimerizedsplit Cas9系统
,能够控制有活性Cas9的产生,从而降低脱靶效应。最后通过Cas9中几个氨基酸的突变也能够提高DNA的特异性。
在spCas9中,
HF Cas9(N497A/R661A/Q695A/Q926A)与eCas9(K848A/K1003A/R1060A)能够中和蛋白与DNA磷酸糖骨架之间的非特异性静电作用
,从而显著提高Cas9对DNA的特异性。
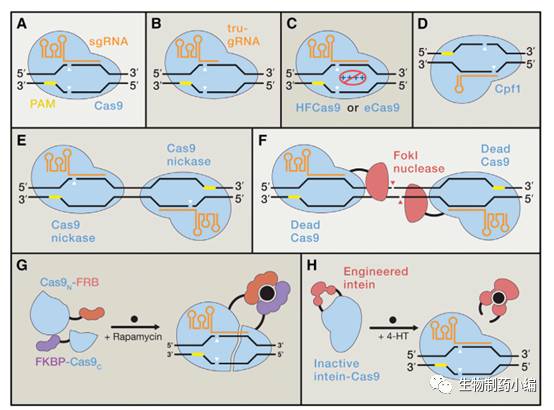
▲
提高
CRIPSR
系统
DNA
特异性的方法
精准编辑(
precise editing
)
。
由于同源依赖的修复
HDR
效率很低(
<5%
),大部分由
Cas9/Cpf1
产生的
DNA
双键断裂都由
NHEJ
修复完成,从而会引进许多随机的插入和缺失。此外,
HDR
的效率还与细胞的类型与状态等因素相关,因此如何提高
HDR
效率从而完成基因的精准编辑也是
CRIPSR
技术用于临床的巨大挑战。
其中一个做法是
使用单个
Cas9n
与
donorDNA
底物
,由于
DNA
缺口基本不会导致
NHEJ
,因此此方法只产生很少的插入和缺失,
但是其编辑效率与野生型相比显著下降,且能应用的细胞类型也十分有限
。由于
HDR
与
NHEJ
的竞争关系,
使用小分子的
NHEJ
抑制剂或者
HDR
的增强剂也能够提高
HDR
的发生频率
。
DNA ligase IV
的抑制剂
Scr7
,
DNA-PKcs
抑制剂,与
KU70/KU80
的抑制剂或者
Rad51
的小分子激活剂都能够提高
HDR
介导的基因编辑频率。此外,
将细胞同步在
G-phase
也能够提高
HDR
频率
。但是这些小分子抑制剂可能会影响细胞正常的功能,因此在实际使用中会有诸多限制。
同时在同源
DNA
模板上也能做一些改进。由于切割完成后,
Cas9
从
DNA
上的解离是不对称的。若同源模板
DNA
能与最先被释放出的
DNA
退火的话,
HDR
介导的基因编辑效率就会提高。为了避免完成编辑的
DNA
再次被
Cas9
识别切割,
可以通过突变同源模板
DNA
,使得
HDR
产物形成突变的
PAM
。
碱基编辑(
baseediting
)
是一种引入点突变的新策略,它不依赖于
HDR
或者
DNA
双键断裂。其主要方法是融合失活的
Cas9
(
dCas9
)与一个胞嘧啶脱氨酶,在
dCas9
,
sgRNA
和目的
DNA
互补链形成三元复合物时,胞嘧啶脱氨酶能催化非互补链(单链)上的
C
(互补序列
3-5
个碱基位置处)转化为
U
,接着引发细胞错配修复从而将原先的
C:G
替换为
T:A
。这种方法的编辑效率比
HDR
介导的点突变更高,且插入缺失突变更少。但是仍存在一些不足:首先需要被编辑的
C
距离
PAM
序列特定的位置,限制了广泛应用;其次可能无法分辨编辑窗口中的多个
C
,从而特异性降低。
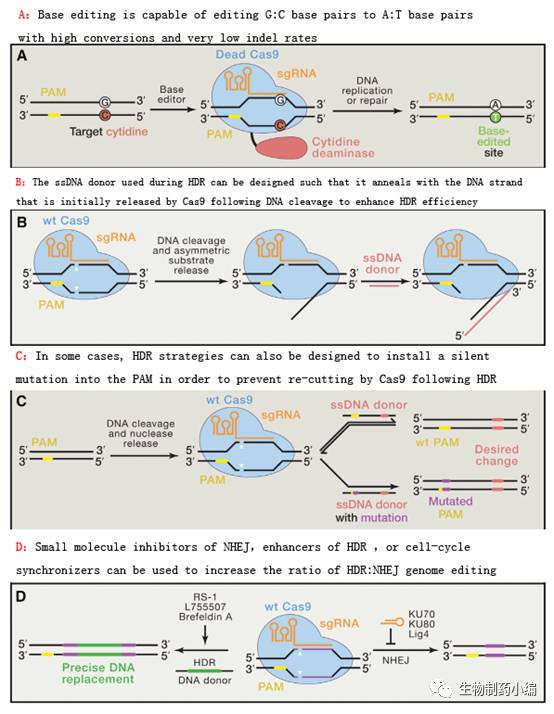 ▲
提高精准基因编辑效率的办法
▲
提高精准基因编辑效率的办法
CRISPRa
与
CRISPRi
。
CRIPSR
除了用于精准编辑某个基因,还能用于调控基因的表达。当将转录激活结构域
VP64
与
dCas9:sgRNA
融合,就能够激活特定基因的表达。第二代
dCas9-activator
融合蛋白使用了多个拷贝的
VP16
,三元激活因子
VPR
(
VP64-p65-Rta
)或者串联重复的
VP64
表位标签肽段。此外,转录激活因子
MS2-p65-HSF1
等也可以通过结合
sgRNA
上的
RNA
发卡结构从而实现基因的转录激活。同时使用
dCas9-VP64
与
MS2-p65-HSF1
,被称为协同激活调控(
synergistic activation mediator(SAM)
),展现出了超强的转录激活能力。相反的,失活的
Cas9
(
dCas9
)本身或者
dCas9
融合转录抑制因子
KRAB
能够抑制特定基因的表达。而且,上文提到过的
small-molecule-activateddCas9
融合
VP64
或
KRAB
,就能够实现特定基因时空特异地激活与抑制。
除了直接融合转录激活
/
抑制因子,
dCas9
还可以融合相关的表观遗传因子。如
dCas9
融合甲基转移酶
DNMT3A
使目标
DNA
区域
100bp
范围内甲基化水平升高;而
dCas9
融合去甲基化酶
Tet1
能使目标
DNA 200bp
范围内发生去甲基化。
dCas9
与
H3K27
乙酰化酶
p300
或者组蛋白去甲基化酶
LSD1
的融合,能够影响目的区域近千
bp
的组蛋白修饰变化,从而实现基因的转录调控。
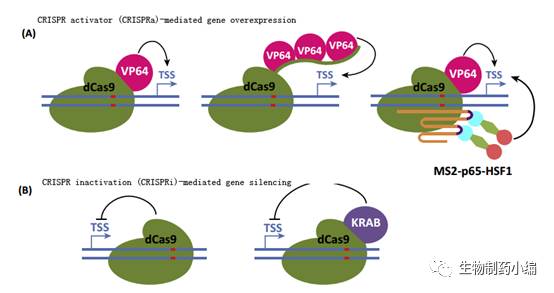
▲
CRISPR
介导的基因转录激活与抑制
基因编辑试剂的递送(
delivery
)。
由于相关的基因编辑试剂都是蛋白,核酸等大分子,不能够自发地进入细胞内。此外,在血液中裸露的核酸还会被核酸酶降解从而引发免疫反应。因此基因编辑试剂如何能克服这些障碍,从而进入细胞核发挥作用是实现体内基因编辑的一大难题。
目前用于体内基因编辑递送主要有以下方法:
高压注射法(
Hydrodynamicinjection ,HDI
),脂质
-
纳米颗粒递送(
Lipidnanoparticle delivery
,
LNP
)以及依赖病毒载体的递送(
Viraldelivery
)
。在老鼠乙肝病毒模型中,
HDI
递送的
Cas9
和
sgRNA
质粒能够有效的降低乙肝病毒的表达量,但是在人临床实验中,
HDI
的应用被其毒性和低转换效率所限制。利用
Lipid nanoparticle
,可以将纯化好的
Cas9:sgRNA
核苷酸蛋白复合物(
RNP
)递送到细胞内。与递送
mRNA
或者
DNA
质粒相比,递送
RNPs
中
Cas9
蛋白发挥活性的时间窗口更短,从而提高特异性,降低脱靶发生的频率。目前有多项利用
LNP
技术进行的基因编辑产品正在开发之中。
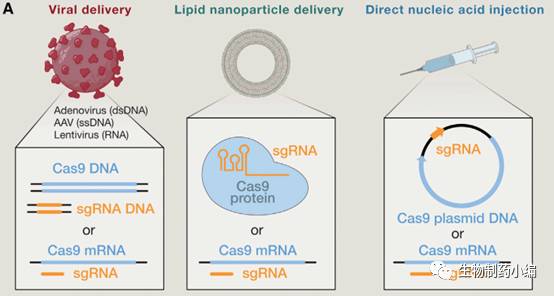
▲
用于体内基因编辑的方法
曾在
90
年代用于基因治疗的病毒载体主要存在两个问题,一是质粒被整合到了基因组中错误的位置,导致了致癌基因的激活;而是因为病毒载体存在的高免疫原性,引发了宿主强烈的免疫反应,导致多器官功能衰竭与脑死亡。经过了近
20
年的改进,现在的病毒载体显示出更高的转化效率与更好的安全性质。现在主要使用的病毒载体包括
慢病毒(
lentivirus
),腺病毒(
adenovirus
)和腺相关病毒(
adeno-associatedvirus, AAV
)
。慢病毒是逆转录病毒的一种,它能够将病毒
DNA
整合到不分裂的细胞中。为了避免错误整合而导致的致癌基因激活,整合酶缺陷的慢病毒载体(
Integrase-defectivelentiviral vectors, IDLVs
)也被开发出来。慢病毒能够包装多达
8.5 kb
的
DNA
,足够满足
Cas9
,
sgRNA
和其他调控元件的包装。腺病毒能够感染分裂和不分裂期的细胞,而不整合进基因组。
然而腺病毒有很高的免疫原性可能会引发很强的免疫反应
。腺相关病毒来自细小病毒家族,是目前发现的一类结构最简单的单链
DNA
缺陷型病毒。
AAV
也能够感染分裂和不分裂期的细胞,而不整合进基因组;同时由于
AAV
在人体中广泛存在,所以其免疫原性也非常低。
此外,由于
AAV
有多种血清型,它可以用于器官特异性的递送
。
AAV
载体所面临的最大问题是它的包装限制为
4.5kb
,而最常用的
SpCas9
的大小为
4.2 kb
,几乎不可能将基因编辑的所有原件组装进一个载体。改进方法之一是使用更小的
Cas9
,如
SaCas9
大小为
3.2 kb
。另一个方法是使用两个
AAV
载体分别包装一半
split-intein Cas9
,当两个
AAV
载体共转时能形成完整功能的
Cas9
从而进行基因编辑。由宾夕法尼亚大学发明的
AAV2.0
技术授权给了
REGENXBIO
进行商业化开发,目前超过
70%
的临床
AAV
项目都采用了此平台。
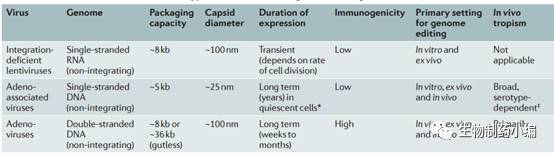 ▲
慢病毒,腺病毒与腺相关病毒的特点
▲
慢病毒,腺病毒与腺相关病毒的特点
CRIPSR
相关公司
。小编之前的文章《
基因编辑能走多远:
CRISPR
引领的精准医疗
》中已经详细介绍了张锋创办的
Editas Medicine
;
Jennifer Doudna
的
Intellia Therapeutics
和
Emmanuelle Charpentier
等人联合创办的药物研发公司
CRISPR Therapeutics
。其中
Intellia Therapeutics
主要使用的递送方法是
LNP
,而
Editas Medicine
同时使用了
LNP
和
AVV
两种方法。在中国,华西医院率先开展世界首个
CRISPR
临床试验。试验采用
ex vivo
的方法,利用
CRISPR
技术敲除患者
T
细胞中的
PD-1
,用于治疗用于治疗非小细胞肺癌。这次临床试验主要是为了检测这项疗法的安全性,希望能早日看到该实验的进展。
CRISPR
与靶点发现
CRIPSR/Cas9
除了直接用于基因编辑,也能够用于药物靶点的大规模筛选。现有的
shRNA
文库筛选存在两个缺点:一是
RNAi
不能够做到完全
knockdown
,因此会有很多假阴性;二是
RNAi
脱靶频率高,会导致很多的假阳性。
CRISPR
技术可以减少这些干扰,同时由于
sgRNA
序列一般比较短,可以实现高通量的
array-based
寡核苷酸合成,从而构建
sgRNA
文库。
CRISPRi LOF
(
loss-of-function
)文库与
CRISPRa GOF
(
gain-of-function
)文库都可以以细胞生长为指标,大规模地分析癌细胞中的遗传依赖关系。此外,在加入某药品的情况下进行筛选,可以分析该药物产生耐药性的机制。比如在黑色素瘤细胞系
A375
中进行
BRAF
抑制剂
vemurafenib
的耐药性筛选发现,肿瘤抑制因子
NF2
,
culin E3
连接酶
CUL3
以及一些组蛋白乙酰转移酶
STAGA
复合物成员的缺失,会导致
vemurafenib
耐药性产生。
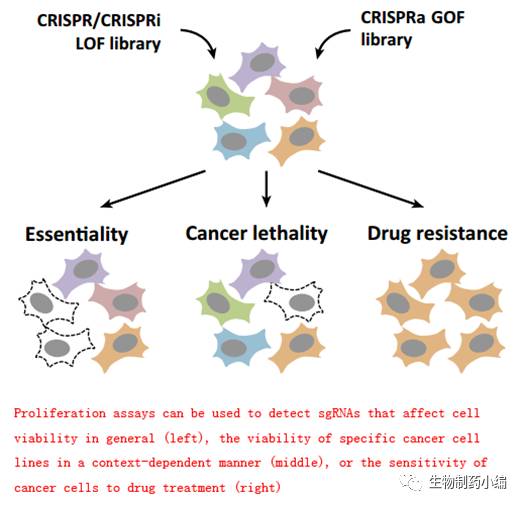
▲
利用
CRISPRi
和
CRISPRa
进行高通量的药物靶点筛选
同样的,
CRISPR
也能够用于最近异常火爆的“协同致死(
synthetic lethality
)”基因的筛选。最近《
Nature Methods
》上发表了
Trey Ideker
和
Prashant Mali
教授的工作,他们利用
CRISPR
技术,在
HeLa
,
A549
和
293T
三个细胞系中,发现
73
个癌基因与药物靶点之间,存在着
152
对协同致死效应组合。其中包括
28
对也被证实的组合,如
BRCA1-PARP1
和
PTEN-MTOR
。通过药物进一步确认,这个结果的准确精确性在
75%
以上,因此能用于大规模筛选协同致死基因。
Dual-gRNA
文库是通过
array-based
寡核苷酸合成建立的,涵盖了
10
万多组基因组合。因此每个构建包含两个
gRNA
,靶向两个目的基因,根据其对细胞生长的影响从而判断两个基因之间的遗传关系。在所有的
152
对组合中只有
16
组(
10.5%
)是其中两个细胞系共有的,而且不存在三个细胞系共有的组合。这说明了协同致死效应存在着细胞特异性。
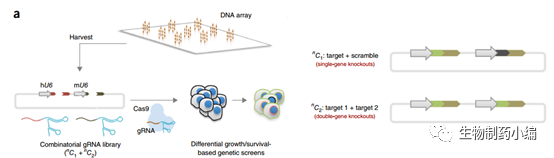
▲
CRISPR
方法筛选协同致死基因组合
C2c2
与与核酸检测
Type VI
家族的
C2c2
于
2016
年被报道,与
Cas9
和
Cpf1
切割双链
DNA
不同,
C2c2
是一个
RNA-guided
的
ssRNA
切割酶。当时作者提出
C2c2
可以用于核酸检测,在今年
4
月的《
Science
》上,张峰课题组带来了
C2c2
的重磅应用,用于检测核酸,其灵敏度可达到阿摩尔级(
aM, 10
的负
18
次方摩尔每升),甚至有望检测出单个核酸,这对于某些诊断的应用有着重要意义。为了得到更为灵敏的信号,选择了
RNase
活性更强的
LwC2c2
。同时为了进一步提高检测灵敏度,也使用了“等温扩增”(
isothermal amplification
)技术。待检
DNA
经重组聚合酶(
recombinase polymeraseamplification, RPA
)扩增后再经
T7
转录为
RNA
,当
LwC2c2
识别这
RNA
后,就可以发挥非特异的
RNase
活性,从而将系统里添加的
RNA
荧光探针切开,释放出荧光。此方法的灵敏度达到了单分子级别,且能够用于癌症液体活检等。由于这套系统中的成分都可以采用冻干粉的方法保存,有利于实现这套系统的商业化。
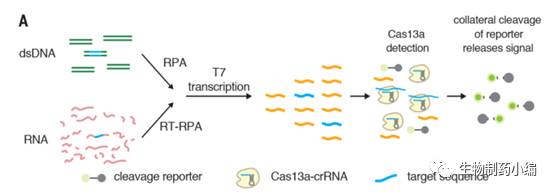
▲
C2c2
用于核酸检测示意图
争议!
CRISPR
的脱靶效应
虽然上文已经介绍过多种方法用于降低
CRISPR
的脱靶效应,但是今年
5
月《
Nature Methods
》上发表了一篇论文,文章发现在在小鼠的体内实验中,
CRISPR
技术会引入数百种意料外的基因突变。研究者们对两只接受了
CRISPR
基因编辑的小鼠和一直未接受编辑的小鼠进行了全基因组测序,结果发现两只小鼠中都出现了
100
多种插入
/
缺失突变,而对照组小鼠只有
3-4
种。令人诧异的是在两只小鼠中,有
68%
的突变是两者共有的,且这些突变的基因组序列与
sgRNA
序列之间的同源性并不高。这篇研究迅速成为了整个生物圈关注的热点,也有科学家对此结果提出了质疑。
Editas Medicine
的首席技术官
Vic E. Myer
与著名科学家
GM Church
联合发文,他们认为研究的数量非常有限,可能会限制观察统计的重现性和可靠性。此外作者不能排除实验动物和单个对照之间报告的基因组差异在
CRISPR
实验操作之前存在的可能性。
任何技术的发展总会遇到波折,希望这样的学术探讨能够促进对
CRISPR
技术特异性的理解,使得
CRISPR
药物早日进入市场,造福人类。
参考文献
-
CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes, Cell, 2017
-
CRISPR/Cas9:From Genome Engineering to Cancer Drug Discovery, Trends in Cancer, 2016
-
SnapShot: Class 2 CRISPR-Cas Systems, Cell, 2016
-
Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems,Science, 2016
-
Delivery technologies for genome editing, NATURE REVIEWS | DRUG DISCOVERY, 2017
-
Combinatorial CRISPR–Cas9 screens for de novo mapping of genetic interactions, Nature Methods, 2017
-
Nucleic acid detection with CRISPR-Cas13a/C2c2, Science, 2017
-
CRISPR gene editing can cause hundreds of unintended mutations, Nature Methods, 2017
扫描下图二维码,您可以直接给本文作者At.Zhou博士打赏。
欢迎加入小编团队成为小编一员
请加小编微信号:wuwenjun7237
如有技术解读、行业洞见愿意分享
欢迎投稿到小编邮箱:
[email protected]

版权为生物制药小编所有。欢迎个人转发分享。其他任何媒体、网站如需转载或引用本网版权所有内容须获得授权且在醒目位置处注明“转自:生物制药小编”。

坚持原创、坚持专业
欢迎关注
生物制药小编
投稿信箱:[email protected]
小编团队现有13位成员:
Armstrong、医药局外人、Fairy、
Jone、
东胜西牛、Alpharesearcher、MT、百草、
Irene、北望、蛋白工人、At.Zhou、Julia
欢迎有共同兴趣的朋友加入





