分子印迹技术是近几十年发展起来的一门集分子设计、高分子合成、分子识别、仿生生物工程等众多学科优势发展起来的交叉学科分支,在手性药物拆分、化学仿生传感器、模拟抗体、模拟酶催化、膜分离、结构类似物的分离、固相萃取以及临床药物分析等领域展现了良好的应用前景。分子印迹聚合物的特异性识别原理是当模板分子(亦称印迹分子)与聚合物单体接触时会形成具有多重作用位点的复合物,经单体聚合并除去模板分子后,聚合物中的空穴与模板分子空间构型相匹配且能与模板分子再次形成多重作用位点。因此,该空穴将对模板分子及其类似物具有选择识别特性,即具有记忆性。
目前,根据模板分子和聚合物单体之间作用方式的不同,分子印迹技术主要分为
3
种类型
[1-2]
:(
1
)共价键型。先将模板分子(或印迹分子)与功能单体发生缩合反应、酯化反应等反应得到相对稳定的中间体(如硼酸酯、亚胺等),再经交联聚合得到相应
的高聚物,最后利用水解等化学反应脱掉模板
分子
(
或印迹分子
)
制备出对应的共价键型分子印迹聚合物。(
2
)非共价键型。单体与模板分子之间的作用方式主要包括静电引力、与金属离子形成的配位键、氢键作用力、芳香环间的
π-π
作用力、较大疏水基团之间的范德华力等超分子作用力。其中最具典型的是离子静电引力和氢键作用力。由于非共价键型分子印迹技术其聚合物制备条件温和、模板分子抽提容易,因此是目前最常用的方法。(
3
)半共价键型。在预聚过程中功能单体与模板分子以共价键形成复合物,交联聚合后脱掉模板分子,但在识别过程中功能单体与模板分子以非共价键结合,该方法也被称为“牺牲空间法”。
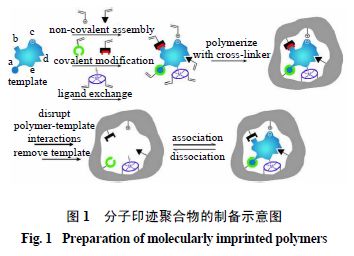
在制备分子印迹聚合物过程中,首先模板分子与功能单体依靠官能团之间的共价或非共价作用形成主客体复合物。然后通过交联剂交联聚合或者引发剂引发单体聚合,形成高交联的刚性聚合物,最后将模板分子洗脱或解离得到分子印迹聚合物。制备分子印迹聚合物所采用的聚合方法主要有本体聚合法、沉淀聚合法、悬浮聚合法、乳液聚合法、原位聚合法、电聚合法以及表面印迹聚合法等方法。分子印迹聚合物的制备过程见图
1
。
分子印迹聚合物材料主要有不规则的研磨颗粒、规则的球形颗粒、纳米颗粒、毛细管柱、位于毛细管内的开口管层、薄层表面、印迹膜以及复合物等形式。分子印迹技术应用领域包括食品分析、样品前处理、传感器、酶催化模拟、天然产物分离、手性拆分以及免疫分析等众多领域,具有广阔的发展空间。
中药材化学组分复杂,其活性成分纯化方法主要有固相析出分离法(盐析沉淀法
[3]
、有机溶剂沉淀法等)、分子蒸馏技术、吸附分离技术
[4]
、膜分离、电离分离技术等。为了获得较高纯度的活性成分,使用最为广泛的是吸附分离法和色谱分离法,包括气固色谱(
GSC
)、液液色谱(
LLC
)、液固色谱(
LSC
)、超临界流体色谱(
SFC
)
[5]
。应用于中药活性成分分离的材料(常见的有离子交换树脂、吸附树脂、硅胶、渗透膜以及氧化铝等)由于对目标产物的吸附缺少选择性,通常需要多次柱色谱以及反复重结晶等操作才能获得较高纯度的活性成分,工艺复杂、分离周期长、效率低,而且大量的溶剂消耗和吸附材料(难降解)的丢弃会造成严重的环境污染。而分子印迹技术具有制备简单、成本廉价、能重复使用等优点,尤其是特异的分子识别性能、高效率的富集与分离能力能够使结构类似的物质很容易得到分离,使中药成分纯化工艺得以简化,节省了溶剂的用量,减少了环境污染,因此分子印迹聚合物将会在中药活性成分的分离纯化应用中具有极大的发展空间。影响印迹聚合物分离效果的因素主要有功能单体的结构、聚合方法、交联剂的选择以及印迹材料具体存在形式等因素,其中功能单体自身结构与模板分子结构之间的匹配性和结合作用的强弱是决定印迹聚合物分离效果的决定性因素。本文基于中药活性成分的种类综述分子印迹技术富集与分离中药活性成分的应用进展,尤其是对分子印迹聚合物的构建、单体交联剂的选择、对结构类似物的分离效果,以及从中药材提取液中萃取模板分子等方面进行总结,以期为后续研究中设计结构新颖、分离效果更好的印迹聚合物提供参考。
1
黄酮类成分
黄酮类化合物是基本母核为
2-
苯基色原酮类化合物(包括黄酮、异黄酮、新黄酮、黄酮苷等)。利用分子技术分离纯化黄酮类化合物可基于聚合单体(含酰胺基团、吡啶环等)与黄酮结构中的酚羟基之间形成的特异性氢键识别力来富集分离出黄酮化合物,也可以基于功能单体中芳香环与黄酮类化合物的
π-π
作用力或者范德华作用力来特异性识别并分离出黄酮类化合物。
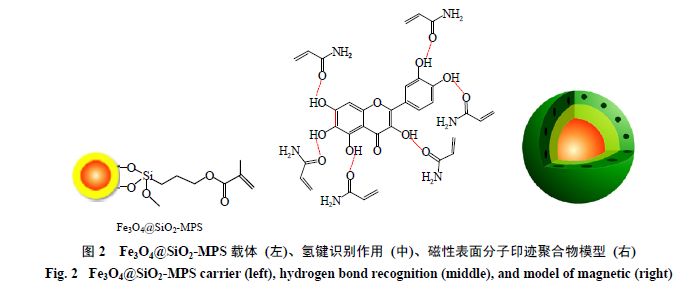
Ma
等
[6]
以超顺磁核壳结构的
Fe
3
O
4
@SiO
2
为载体(图
2
),丙烯酰胺作为功能单体,乙腈为致孔剂,合成出栎草亭
(
quercetagetin
)
磁性表面分子印迹聚合物(图
2
)。该聚合物对栎草亭的最大吸附量为
22.65 μmol/g
,解离常数为
236.97 mL/g
,栎草亭相对结构类似物木犀草素、牡荆苷、芦丁和白藜芦醇的选择性系数分别为
1.50
、
2.67
、
4.74
和
4.74
。使用该
分子印迹聚合物能成功从金盏花提取物中
分离栎草亭。与高效液相色谱
-
紫外联用法
(
HPLC-UV
)
和高效
液相色谱
-
质谱联用法(
HPLC-MS
)等方法相比,印迹技术显示出更高的选择性和时效性。由于栎草亭极性较大,若使用强极性的溶剂容易削弱模板分子与功能单体之间的氢键作用(影响聚合物分子识别性能,图
2
),因此使用弱极性的乙腈作为溶剂和致孔剂。
Zhang
等
[7]
以
3-
三乙氧基甲硅烷基
-1-
丙胺修饰过的
SiO
2
为载体,以甲基丙烯酸为功能单体制备了表儿茶素没食子酸酯(
EGCG
)表面分子印迹聚合物,该聚合物对
EGCG
的最大吸附量为
0.55 mg/g
,分布系数高达
22.5
。使用甲醇
-
乙酸(
7
∶
3
)洗脱液,该聚合物能从茶多酚溶液中选择性地富集与分离出
EGCG
,不受
(+)-
儿茶酚(
C
)、
(−)-
表儿茶酸(
EC
)、
(−)-
表没食子儿茶素(
EGC
)等结构类似物的干扰。买买提
•
吐尔逊等
[8]
以丙烯酸作为功能单体,苄基三硫代碳酸酯作为转移剂,经过可逆加成
-
断裂链转移聚合制备了山柰酚分子印迹整体柱,该方法得到的聚合物相孔径较小,比表面和孔隙率较高,骨架结构均匀。选择性实验得出山柰酚相对结构类似物槲皮素的平均分离因子为
1.52
。谢娟平
[9]
以甲基丙烯酸(
MAA
)为功能单体,乙二醇二甲基丙烯酸酯(
EDMA
)为交联剂,合成了双藿苷
A
分子印迹聚合物并成功从淫羊藿粗提物中固相萃取出双藿苷
A
,质量分数由
10.7%
提高到
59.6%
。王松等
[10]
以
MAA
为功能单体,二甲基丙烯酸乙二醇酯(
EGDMA
)为交联剂,经沉淀聚合法制备了黄芩素分子印迹聚合物。黄芩素和
MAA
单体之间通过氢键自组装形成
1
∶
4
的复合物,相比结构类似物槲皮素和结构非类似物氯霉素,黄芩素的分离因子分别为
17.69
和
26.03
。
在提取工艺的简洁性和活性成分的纯度上,分子印迹技术明显优于超声提取法、酶提取法、超临界流体萃取法、大孔树脂吸附法等常规方法。基于分子印迹技术分离黄酮类化合物国内研究者对芦丁、黄芩素、黄芩苷以及槲皮素等活性成分的研究较多。
邱磊等
[11]
以丙烯酰胺(
AM
)为功能单体,以沉淀聚合法制备了黄芩苷分子印迹聚合物,相对结构类似物汉黄芩苷和黄芩素,该聚合物对黄芩苷的分离因子分别为
1.3
和
1.6
。顾小丽等
[12]
以黄芩苷为模板分子,分别制备了沉淀分子印迹聚合物(
MIP
)、纳米二氧化硅表面分子印迹聚合物(
SiO
2
-MPS
)以及介孔分子筛
SBA-15
表面分子印迹聚合物(
SBA-15-MPS
),最大吸附量分别为
31.84
、
40.84
和
68.29 μmol/g
,分别为相应非分子印迹聚合物的
1.53
、
1.97
和
2.06
倍。
SBA-15-MPS
具有更好的吸附性和亲和性,为黄芩苷的分离提供新材料。王素素等
[13]
以
4-
乙烯基吡啶(
4-VP
)为功能单体,采用本体聚合法制备了芦丁
-
槲皮素复合模板分子印迹固相萃取柱,可从槐米提取物中选择性分离芦丁和槲皮素
2
种黄酮类化合物,总回收率分别为
96.70%
和
94.67%
。阮永欣等
[14]
以
KH570-Fe
3
O
4
为载体,
N
-
乙烯基吡咯烷酮(
NVP
)和
AA
为功能单体,
N
,
N
′-
亚甲基双丙烯酰胺(
MBA
)为交联剂,制备了槲皮素磁性表面分子印迹聚合物(
Qu-MMIPs
),该聚合物对槲皮素以及结构类似物染料木素的分离因子分别为
2.32
和
1.32
,说明对槲皮素具有较高选择性吸附。
此外,由于一些黄酮分子和聚合单体能与金属离子配位形成较稳定的配合物,因此以配合物作为模板分子构建的分子印迹聚合物在金属离子存在下对这些黄酮类化合物具有更高的特异性识别。
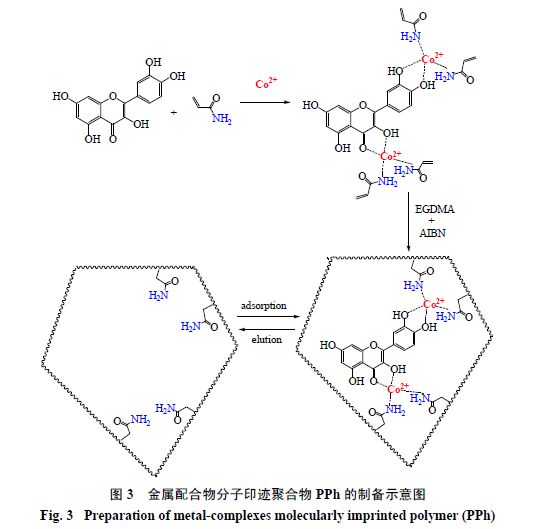
Huang
等
[15]
以槲皮素的钴配合物
[Qu-Co
(
II
)
]
为模板分子,以丙烯酸为聚合单体经自由基引发聚合得到一种金属配合物分子印迹聚合物
PPh
(图
3
),
另外以槲皮素为模板分子制备的印迹聚合物为
PQu-Ph
。吸附热力学表明
PPh
在加入钴离子后对槲皮素的吸附容量大于
PQu-Ph
对槲皮素吸附量,说明在印迹聚合物制备过程中金属配位键相比非共价键(如氢键、范德华力等)作用强。
PPh
对槲皮素的解离系数为
48.73
,对结构类似物芦丁和
4,5,7-
三羟黄烷酮的解离系数分别为
7.54
和
8.75
,槲皮素相对芦丁的分离因子为
6.46
,相对
4,5,7-
三羟黄烷酮的分离因子为
5.56
,该结果表明
PPh
明显优于槲皮素印迹聚合物
PQu-Ph
。成洪达等
[16]
以
MAA
为功能单体,
EGDMA
为交联剂,制备了芦丁
-Cu
2+
配位分子印迹聚合物,该聚合物与芦丁
-Cu
2+
配位分子通过配位键及分子间作用力结合芦丁
-Cu
2+
配位分子,具有特异性识别作用(相对黄芩素的分离因子为
2.53
,相对葛根素的分离因子为
3.17
),可用于中药提取物中芦丁的分离和富集。
综上所述,分离黄酮类化合物的分子印迹聚合物大多属于非共价键型(基于氢键作用或者金属离子配位作用),聚合方法以沉淀聚合法和表面印迹聚合法较多见,同时原位聚合法(如分子印迹整体柱得到的孔径和粒径较均匀,比表面积和孔隙率也较高)和介孔分子筛表面印迹聚合法(与传统表面印迹技术相比,由于介孔内也有识别空穴,因此吸附量更大)具有较大潜在发展优势。
2
生物碱类成分
生物碱指主要来源于植物界的一类含氮有机化合物,由于生物碱具有抗病毒、抗肿瘤、免疫抑制、抗炎、镇痛等多种功效而成为近十几年来的
1
个热点研究对象。许多植物体内高活性的生物碱(如紫杉醇、喜树碱等)量较低,采用传统的色谱分离法难以获得高纯度的产品。基于分子技术分离生物碱,使用最为广泛的是利用生物碱分子结构中的氮原子与聚合单体中的羧基或酰氨等基团之间形成的氢键作用来特异性识别。
Zhang
等
[17]
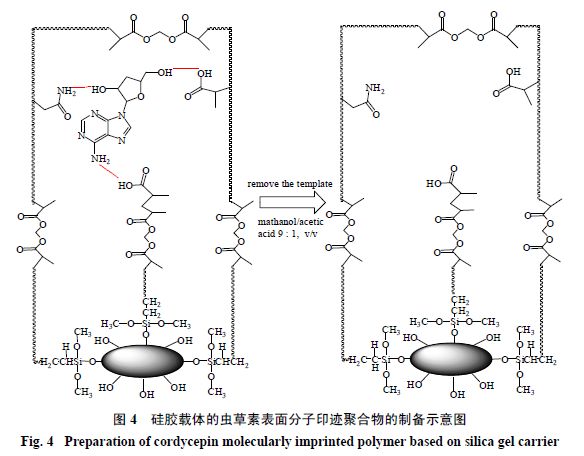
以甲基丙烯酸和丙烯酰胺为聚合单体,以乙烯基三乙氧基硅烷修饰过的硅胶为载体,制备了虫草素表面分子印迹聚合物(图
4
),该聚合物对虫草素的吸附量为
95.37 mg/g
。相对次黄嘌呤核苷,虫草素吸附选择性系数为
3.14
;相对腺苷,虫草素吸附选择性系数为
3.74
。由该聚合物制备的固相萃取柱从北冬虫夏草提取液中选择性萃取出虫草素的质量分数高达
98%
,回收率为
25.67%
。
Fan
等
[18]
以
Fe
3
O
4
@GO
复合物纳米颗粒为载体,
吴茱萸碱为模板分子,甲基丙烯酸(
MMA
)为聚合单体,制备了吴茱萸碱磁性表面分子印迹聚合物
MIP@Fe
3
O
4
@GO
(图
5
),该聚合物兼具吸附选择性高且制备容易(源自
MIPs
)、表面修饰方便(源自氧化石墨烯
GO
)、从母液中去除容易(源自磁性
Fe
3
O
4
,相比其他载体,无需离心或滤过,仅用磁铁就能去除)三重优势。该聚合物对吴茱萸碱的最大吸附量为
15.581 9 mg/g
,从吴茱萸提取液中能选择性富集与分离出吴茱萸碱和吴茱萸次碱,回收率分别为
82.17%
和
68.24%
。
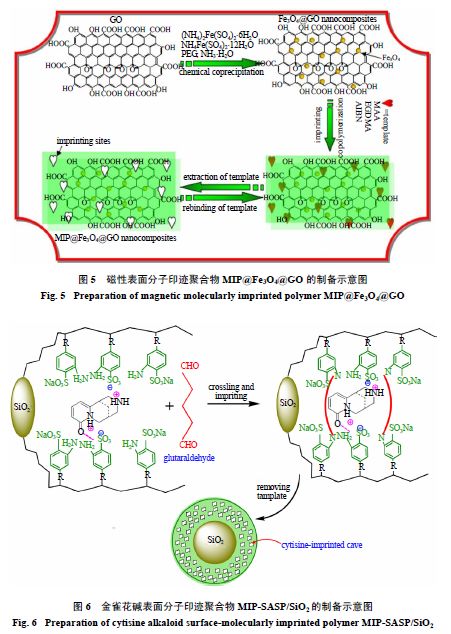
Gao
等
[19]
先将甲基丙烯酸缩水甘油酯(
GMA
)接枝到硅胶微粒上得到
PGMA/SiO
2
接枝微粒,随后与
2,4-
二氨基苯磺酸钠反应,发生环氧开环缩合得到
SAS-PGMA/SiO
2
功能接枝微粒。最后以金雀花碱为模板分子,戊二醛为交联剂,制备出了金雀花碱表面分子印迹聚合物
MIP-SASP/SiO
2
(图
6
)。相对于苦参碱和氧化苦参碱
2
种结构类似物,该聚合物对金雀花碱的选择性系数分别为
9.5
和
6.5
。随后
Gao
等
[20]
又将氯甲基化的聚砜(
CMPSF
)经氨基修饰得到氨基化的聚砜膜(
AMPSF
),再经甲基丙烯酸接枝得到接枝聚砜膜(
PSF-g-PMAA
)。最后将该膜与交联剂乙二醇二缩水甘油醚(
EGDE
)共聚得到苦参碱嫁接型分子印迹膜(
GMIM
)。该印迹膜是由薄的印迹层、纳米级别的通道和具有大孔结构的支撑层组成,对苦参碱具有高的分子识别选择性、良好的亲和力和选择渗透性。苦参碱相对结构类似物金雀花碱的分离系数达到
5.9
。
曾绍梅等
[21]
以甲基丙烯酸缩水甘油酯(
MAA
)为功能单体,三羟甲基丙烷三甲基丙烯酸酯(
TRIM
)为交联剂,合成了樟柳碱(
ASD
)分子印迹聚合物,该聚合物对
4
种托品烷类生物碱(樟柳碱、东莨菪碱、山莨菪碱、阿托品)具有类特异性识别能力,能够富集、分离和检测马尿泡果实中
4
种生物碱,回收率为
70.0%
~
96.3%
,
RSD
<
5.7%
。朱俊访等
[22]
通过光聚合方法制备了左旋延胡索乙素分子印迹聚合物,并研究了其特异性吸附性能。吕建峰等
[23]
采用聚酰亚胺纳米纤维膜表面预涂覆聚甲基丙烯酸,利用
EDMA
进行交联聚合制备了茶碱分子印迹纳米纤维复合膜。该膜对茶碱的静态吸附结合容量达
144 μmol/g
,茶碱相对可可碱的分离因子达
1.96
。
紫杉醇作为抗乳腺癌、卵巢癌等癌症的一线临床药物,探索其高效分离方法一直是近几年来研究的热点内容之一。
Li
等
[24]
以
2-
乙烯基吡啶作为功能单体,以马来松香乙二醇丙烯酯作为交联剂合成了紫杉醇分子印迹聚合物,并将该聚合物做成高效液相色谱固定相来分离紫杉醇及其类似物紫杉萜。
结果表明紫杉醇印迹因子达到
2.37
,相对紫杉萜的
分离因子达到
2.54
。闫艳等
[25]
以
MAA
为功能单体,丙酮为致孔剂合成了紫杉醇分子印迹聚合物,经过该聚合物固相萃取后,真菌发酵液中紫杉醇的量显著提高,可为复杂样品中紫杉醇的快速分离纯化提供新材料。利用廉价的优势资源开发出能够提高印迹效率的新型交联剂,对于未来绿色印迹新材料的开发和环境保护具有现实意义。
此外,对于结构较复杂、价格相对较高的活性成分的分离可采用含相似结构片段的廉价分子作为假模板分子制备成相应的印迹聚合物再进行富集与分离。
Lian
等
[26]
采用双模板分子策略,以咖啡因和己酮可可碱为假模板分子合成出了分子印迹聚合物微球并制备成固相萃取柱,结合高效液相色谱荧光检测器(
HPLC-FLD
)可从微小亚历山大藻中提取出膝沟藻毒素
1
、
4
(
gonyautoxin 1
,
4
),提取率为
73.2%
~
81.5%
。
Xia
等
[27]
以聚乙二醇(
PEG
)修饰过的
Fe
3
O
4
为载体,甲基丙烯酰胺(
MAC
)为功能单体,双氢可待因为假模板分子,制备了磁性假模板分子印迹聚合物,该聚合物对吗啡(
MO
)及其类似物曲马多、双氢可待因和加替沙星均有特异性吸附作用,尤其是对
MO
印迹因子达到
2.10
。
在分子印迹聚合物的众多聚合方法中,表面分子印迹法在分离生物碱类化合物应用中最为广泛,其功能单体的选择大多为酸性功能单体(如丙烯酸、甲基丙烯酸等)和弱碱性功能单体(如丙烯酰胺是基于氢键作用力来识别)。
3
苯丙素类成分
苯丙素类化合物是基本母核具有
1
个或者几个
C
6
-C
3
单元的天然有机化合物类群,具有抗血凝、抗病毒、抗骨质疏松以及抗炎等多方面的生理活性。利用分子印迹技术分离苯丙素类化合物,大多是基于功能单体与模板分子官能团之间的氢键以及范德华力等超分子作用力原理来实现特异性分离,其中研究较多的是苯丙酸类(阿魏酸、绿原酸、咖啡酸等)和香豆素类等活性成分的富集与分离。
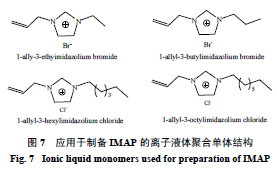
Bi
等
[28]
以氯化
1-
烯丙基
-3-
乙基咪唑鎓盐离子液体为功能单体,阿魏酸为模板分子,乙醇
-
水(
9
∶
1
)为致孔剂,经自由基引发聚合得到基于离子液体的酚酸分子印迹离子交换聚合物(
IMAP
,图
7
)。利用该聚合物可从海蓬子
Salicornia herbacea
L.
提取液中萃取出
3
种酚酸类化合物原儿茶酸(
PA
,回收率
90.1%
)、阿魏酸(
FA
,回收率
95.5%
)、咖啡酸(
CA
,回收率
96.6%
),此外,这
3
种酚酸经过重复性的固相萃取循环过程能够逐一分开。
Miura
等
[29]
以
4-
乙烯基吡啶和甲基丙烯酰胺为单体,二乙烯基苯作为交联剂,乙腈
-
甲苯(
3
∶
1
)作为致孔剂,制备出咖啡酸分子印迹聚合物。亲水作用色谱模式下印迹聚合物主要基于亲水作用力而显示分子识别性能,但在反向分配色谱模式下,印迹聚合物主要是基于氢键和疏水作用力来识别咖啡酸模板分子。在亲水作用色谱模式下该聚合物对绿原酸、五倍子酸、原儿茶酸、香草酸没有识别性能,利用该印迹聚合物成功从杜仲提取液中萃取出绿原酸。
Fan
等
[30]
以
Fe
3
O
4
@SiO
2
为载体,
4-
乙烯基吡啶(
4-VP
)为功能单体,制备了咖啡酸分子印迹聚合物,该聚合物对
CA
具有特异性吸附,其吸附量依次是结构类似物对香豆酸(
pCA
)、
FA
、肉桂酸(
CMA
)和原儿茶酸(
PCA
)的
1.64
、
2.22
、
5.36
和
35.87
倍,该聚合物为从复杂体系中分离富集
CA
提供了新材料。王素素等
[31]
采用
Zn
2+
-
阿魏酸
-
咖啡酸配合物作为模板分子,以
4-VP
为功能单体制备了双分子印迹聚合物,由该聚合物制备的固相萃取柱能够选择性地从升麻样品液中提取出
FA
和
CA
,回收率分别达到
92.7%
和
95.4%
。
Miura
等
[32]
以甲基丙烯酸为功能单体,二乙烯基苯作为交联剂,制备了绿原酸分子印迹聚合物,该聚合物对绿原酸具有特异性吸附作用,而对结构类似物
CA
、没食子酸等没有特异性吸附作用,该聚合物能从杜仲叶中成功分离出绿原酸。
安磊等
[33]
以
L
-
特丁氧羰基苯丙氨酸(
Boc-
L
-Phe
)为模板分子,
MAA
为功能单体,制备了其分子印迹聚合物,将其作为色谱固定相,对其对映异构体进行手性拆分。该聚合物通过协同氢键作用形成
1
∶
3
型配合物,专一地结合
L
-
特丁氧羰基苯丙氨酸分子。董胜强等
[34]
以聚偏氟乙烯(
PDVF
)为载体膜,
MAA
为功能单体制备了
香豆素分子印迹膜,该膜对香豆素的最大吸附量为
0.151
8 mmol/g
,印迹因子达
2.09
,利用该聚合膜从桂枝粗提液中分离富集香豆素,回收率为
89.6%
。廖森
[35]
以氨丙基三乙氧基硅烷和甲基三乙氧基硅烷(
MTEOS
)为双功能单体,
SiO
2
为载体,正硅酸四乙酯(
TEOS
)为交联剂合成去甲二氢愈创木酸(
NDGA
)表面分子印迹聚合物(
NDGA- MIP@SiO
2
)。该聚合物对麻黄乙醇提取液中的
NDGA
吸附容量为
4.90 mg/g
,明显高于非印迹聚合物
NIP@SiO
2
(
1.70 mg/g
),印迹因子高达
2.88
。
对于含羧酸基团的苯丙素类化合物的分离,选择碱性或者弱碱性的功能单体能够增大吸附量有利于提高总回收率;另外,选择代表性结构的具多种活性的成分作为模板分子所制备的印迹聚合物,也能够实现多种模板分子及其结构类似物的交叉选择性识别、分离和富集。
4
糖类和氨基酸类成分
糖类是多羟基醛或多羟基酮及其衍生物、聚合物的总称。使用分子印迹技术分离糖类和氨基酸,可以利用糖分子结构中的羟基与硼酸形成的硼酸酯或者氨基形成的亚胺等共价键首先得到功能单体,再经单体聚合后经水解掉模板分子,即可得到共价型分子印迹聚合物;亦可以通过功能单体与糖(或氨基酸)分子间的氢键、偶极、静电等作用力形成较稳定的复合物,再经单体聚合后抽提出模板分子得到非共价型分子印迹聚合物。
Peng
等
[36]
以
D
-
葡萄糖共价键合的硼酸为聚合单体(硼酸作功能单体、
D
-
葡萄糖为片段模板),以
Fe
3
O
4
固体核为载体,
TEOS
为交联剂(控制壳的厚度)制备出基于硼酸亲合的糖苷表面分子印迹聚合物。该印迹聚合物吸附量大且传质速度快,同时亲水性也得到提高,使用磁铁很容易将其分离。利用该聚合物能对染料木苷(
genistin
)、大豆苷(
daidzin
)、黄豆黄苷(
glycitin
)均显示出特异性吸附,但对芦丁没有特异性吸附作用且吸附量较小,原因是与葡萄糖额外相连接的鼠李糖残余导致的空间效应阻止了芦丁分子进入聚合物空穴。该聚合物结合
HPLC
分析,成功从大豆水提液中萃取出染料木苷、大豆苷和黄豆黄苷,回收率
88.3%
~
105.7%
,
RSD
<
10%
,该方法为苷类识别和具有顺式二醇结构的糖印迹提供了一个通行方法。徐鹏飞
[37]
以
Fe
3
O
4
@SiO
2
@NH
2
@BAD-lectin
为载体,刀豆凝集素和
3-
氨基苯硼酸为双功能单体,
MBA
为交联剂合成了葡萄糖磁性表面分子印迹聚合物。该聚合物微球对葡萄糖的印迹因子为
2.93
,高于鼠李糖(
2.63
)和麦芽糖(
2.19
)。宋亚玲等
[38]
以硅胶为载体,
3-
氨丙基三乙氧基硅烷(
APTS
)为硅胶烷基化改性试剂,五没食子酰葡萄糖(
PGG
)为模板分子,
MAA
为功能单体制备了硅胶表面分子印迹聚合物,利用该聚合物制备的色谱柱能从桂枝茯苓胶囊提取液中分离出
PGG
,质量分数可达
90.2%
。谢宏凯等
[39]
以甘氨酸
-
dl
-
亮氨酸(
GL
)为假模板分子、
4-VP
为功能单体、
MBA
为交联剂、
DMSO
为致孔剂,合成了对三七素具有较好选择识别性的假模板分子印迹聚合物(
DMIPs
)。与结构类似物苯丙酮酸(
PHA
)、
dl
-
酪氨酸(
TYR
)和甘氨酰
-
L
-
苯丙氨酸(
GP
)相比,该聚合物对三七素具有特异性吸附。
Zhu
等
[40]
以乙烯基
-SiO
2
纳米球为载体,
MAA
为功能单体,茶氨酸为模板分子,制备了氨基酸虚拟模板表面分子印迹聚合物,该聚合物对
18
种常见氨基酸具有较大吸附作用且在半小时内达到吸附平衡,印迹因子均大于
3
。
由于糖类化合物分子骨架结构较大,且羟基数量较多,氢键识别选择性较差,为减少非特异性结合位点“包埋”现象,加快物质迁移,使用较多的是表面分子印迹聚合物材料(如磁性
Fe
3
O
4
表面印迹纳米微球、
SiO
2
表面印迹纳米微球等)。另外,可逆加成
-
断裂链转移自由基聚合法等聚合方法能够实现可控活性聚合,具有粒径均匀、形貌较统一、相对分子质量分布窄的特点,提高了分子识别性能。
5
萜类及甾体类化合物
基于分子印迹技术分离不含官能团的萜类及甾体类化合物只能通过母体结构的疏水性与功能单体间的范德华作用力来特异性识别(这种识别作用一般较弱),但对于含功能基团的萜类及甾体类化合物与功能单体间的氢键作用可使分子识别作用进一步增强。
熊晶晶
[41]
以丙烯酰
β-
环糊精和丙烯酰胺为双功能单体,三甲基丙烷三甲基丙烯酸酯为交联剂制备了茶皂素分子印迹聚合物。相对甘草酸和甘草次酸,该聚合物对茶皂素的选择因子分别为
1.16
和
17.18
。基于
POSS
杂化,经可逆加成
-
断裂链转移自由基聚合制备的茶皂素印迹聚合物从茶皂素粗提物分离茶皂素质量分数可达
72.5%
,回收率高达
93.2%
。贾华
[42]
以甲基丙烯酸羟乙酯(
HEMA
)为功能单体,
N
,
N
-
二甲基甲酰胺(
DMF
)为致孔剂制备了甘草酸分子印迹聚合物,由该聚合物制备的
flash
柱能够使甘草提取液中甘草酸的量得到进一步提高。
Berengere
等
[43]
分别以甲基丙烯酸和丙烯酰胺为功能单体,丙烯酸乙二醇酯为交联剂,氯仿为致孔剂,经过热聚合反应合成出白桦脂醇分子印迹聚合物。通过固相萃取(
SPE
)实验得出以甲基丙烯酸为功能单体合成的白桦脂醇分子印迹聚合物显示出更好的吸附选择性,吸附容量为
226 nmol/g
,
回收率高于
70%
,印迹参数达到
17
。由该聚合物制备的固相萃取柱,以甲醇为洗脱液,成功从悬铃木树皮
提取液中分离出白桦脂醇和白桦脂酸。
Chen
等
[44]
以丙烯酰胺为聚合单体,四氢呋喃(
THF
)作为致孔剂,经过热聚合合成出奇任醇分子印迹聚合物(图
8
),丙烯酰胺与白桦脂醇分子之间的识别位点主要是氢键作用。该印迹聚合物对奇任醇的结合位点数为(
1.092
±
0.004
)
mol/g
,加权平均亲和力为(
66.07
±
0.07
)
L/mmol
,明显高于非分子印迹聚合
物
[
(
0.339
±
0.001
)
mol/g