 关注“医械资讯社区”,撑握医疗领域相关的全球法规(如:FDA/CE/CFDA等)动态信息和行业最新资讯、前沿科技等,喜欢我们的文章,记得转发扩散哦~
关注“医械资讯社区”,撑握医疗领域相关的全球法规(如:FDA/CE/CFDA等)动态信息和行业最新资讯、前沿科技等,喜欢我们的文章,记得转发扩散哦~
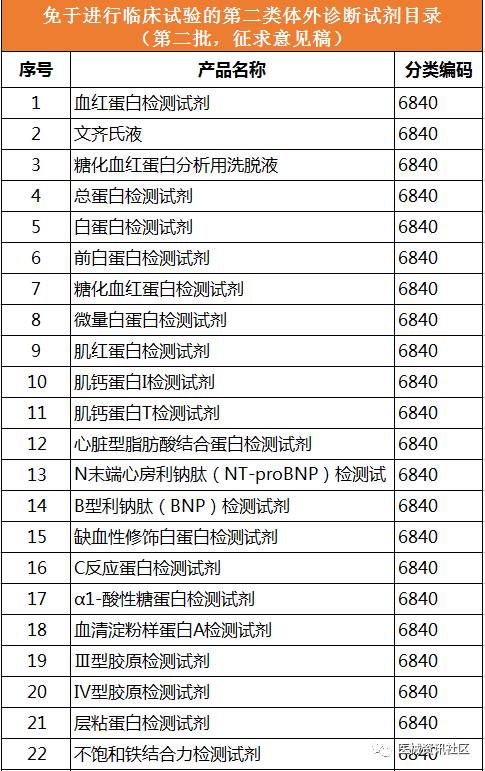
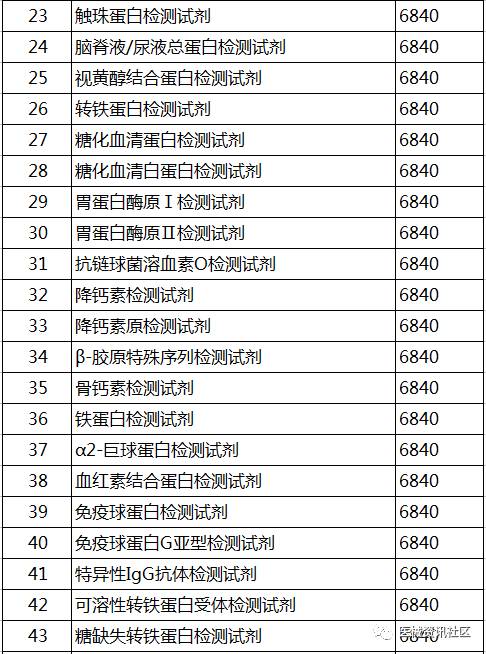
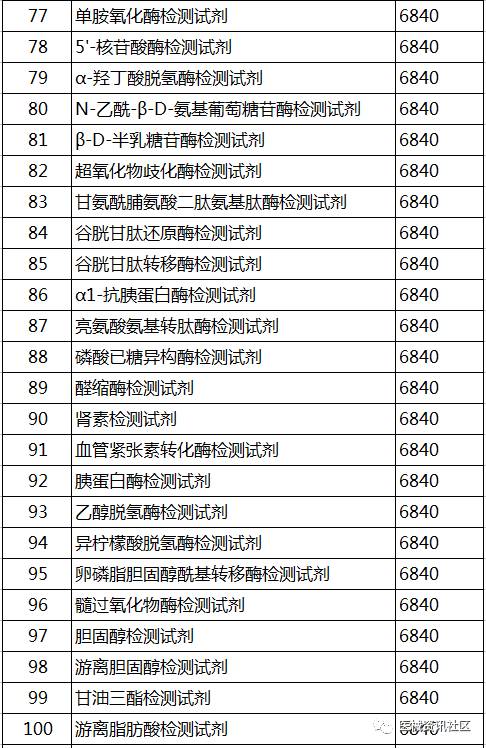
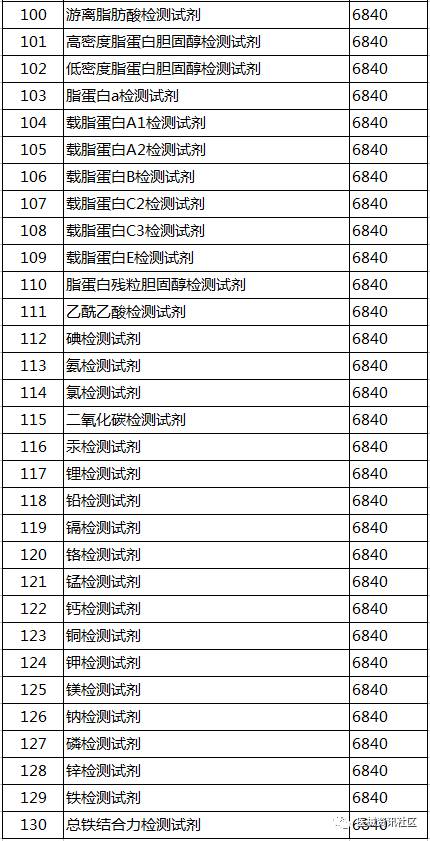
2017年5月24日,CFDA器械注册司组织起草了《免于进行临床试验的第二类体外诊断试剂目录(第二批)》和《免于进行临床试验的体外诊断试剂的临床评价资料基本要求》(征求意见稿,见附件)。要求各省、自治区、直辖市食品药品监督管理局,有关单位在2017年6月30日前反馈意见
电子邮箱:[email protected]。发送邮件时,请在邮件主题处注明“体外诊断试剂相关文件意见”。
信函地址:北京市西城区宣武门西大街26号院2号楼,食品药品监管总局器械注册司,邮编:100053。
其他单位或个人可通过上述电子邮箱或信函地址反馈意见,征求意见截止时间同上。
一、临床评价对比方法
申请人应当根据申报产品的具体情况建立适当的评价方法,充分考虑产品的预期用途,结合产品预期用途开展具有针对性的评价研究。
1.选择已上市产品进行比较研究试验,应选择目前临床普遍认为质量较好的产品作为比对试剂,同时应充分了解所选择产品的技术信息,包括方法学、临床预期用途、主要性能指标、校准品的溯源情况、推荐的阳性判断值或参考区间等,应提供已上市产品的注册信息。
2.选择参考方法进行比较研究试验,应选择参考实验室进行研究,参考方法和参考实验室应具有中国合格评定国家认可委员会(CNAS)认可的资质。
3.根据产品的预期用途也可采用患者的临床诊断、病情进展、疗效观察等客观指标进行临床性能研究。
二、样本选择和样本数量
1.选择涵盖预期用途及干扰因素的临床样本进行评价研究。
2.第二类产品临床病例(样本数量)不少于100例,临床病例应能够充分评价产品临床使用的安全性、有效性。
3.评价用的样本类型应与注册申请保持一致,具有可比性的样本类型如血清、血浆,可选择其中一种样本类型进行临床评价,不具有可比性的样本类型如血清、尿液,应分别进行临床评价。
三、评价报告的基本内容
1.报告封面要求
应包括体外诊断试剂的通用名称、试验开始日期、试验完成日期、试验地点、主要研究者签名及单位盖章、统计学负责人签名及单位盖章、申请人名称(盖章)、申请人的联系人及联系方式、报告日期。
2.试验所用产品信息
包括试验用试剂,对比试剂,配合使用的其他试剂如校准品、质控品、稀释液,试验用仪器等内容,包括具体的试剂名称、生产厂家、规格、批号、失效期,仪器设备应包括名称、生产厂家、型号等内容。
3.评价方案
应包括产品的背景资料、评价目的、评价方法、样本数量、样本类型、人群选择、疾病选择、干扰样本、统计方法、数据处理。
4.评价结果报告
(1)详细描述评价过程中评价方案的执行情况,如具体样本选择情况、试验基本过程等。
(2)试验数据统计结果。
(3)根据试验结果、人群分布、疾病分布、干扰样本、等内容对试验数据进行统计分析。
(4)对于比较研究试验中测定结果不符的样本,应采用合理的方法进行复核,以便对临床试验结果进行分析。如无需复核,应详细说明理由。
(5)试验结论。
5.评价数据表
应至少包括以下内容:样本编号、年龄、性别、试验用试剂检测结果、对比试剂检测结果、复核试验结果(如有)、临床诊断信息(包括干扰样本信息)等。
6.提供试验用产品临床使用情况的相关文献。
7.明确评价资料保存地点为企业自行保管。
四、需注意的问题
1.评价用样本应具有可追溯性。
评价用样本原始资料中应至少包括以下信息:患者样本来源、唯一且可追溯的编号、年龄、性别、科室、临床明确诊断信息、治疗跟踪信息(如有)等内容。
2.注册变更如涉及临床评价应按本要求执行。
3.申请人如无法按上述要求进行临床评价,也可选择临床试验的方式进行临床评价。
征求意见稿目录:

▍来源:CFDA,医械资讯社区原创/整理









