

在药品审评方面,PDUFA(《处方药使用者付费法案》)和GDUFA(《仿制药使用者付费法案》)作为两大主力法案,支撑着美国FDA新药与仿制药的审评与各方面监管活动。一系列使用者付费法案的核心在于通过不同类型的收费法案,合理分配监管资源,建立创新药、仿制药资源互不占用的审评模式,实现创新药和仿制药平衡的双赢局面。
美国以科学、客观的态度去认识药物研发规律,将药品监管活动以科学为基础,并且通过一系列制度设计,前瞻性地把握监管方向,是值得我国借鉴的。
通过CDER和 CBER促进沟通小组(ECT)的内部培训活动,促进FDA在药物开发过程中与临床试验发起人的沟通交流;促进Meta分析方法在药品安全信号检测中的应用;促进生物标记物与药物基因组学的应用;促进以患者报告结局(PROs)和其他终点评价工具的开发;促进罕见病药物的开发。
在新药审评中使用风险效益评价工具;以患者为中心的药物开发项目(PFDD):2016年FDA在银屑病、外周神经病变引发的神经性疼痛、非结核性分枝杆菌感染、器官移植等疾病领域举办了PFDD会议,并对2015年召开的7次疾病相关PFDD会议公布总结报告。
评价风险评估与减低策略(REMS)的实施效果,促进REMS标准化以进一步融入医疗保健体系;通过主动监测体系(即“前哨行动”)开展药品安全性评价;促进药物警戒现代化。
FDA将继续推进强制实施eCTD模式在标准格式化数据提交中的应用,并通过电子提交申请、电子申请数据标准化以及规范临床术语标准,以提高药品审评效率。
eCTD是CDER和CBER从2008年开始推行的提交、修改、补充药品申请的标准化格式,便于FDA审评、批准以及上市后监测药品。
FDA规定,2017年5月5日前,新药上市申请(NDA)、仿制药上市申请(ANDA)、生物许可证申请(BLA)及这些申请的补充申请必须提交eCTD申请。对于新药申请(IND),该时间要求有所推迟,规定到2018年5月5日开始,所有的商业IND申请需要按照eCTD模式提交。在时间节点之后,FDA将不接收任何纸质申请。
PDUFA V提出“促进NME和原创性BLA审评透明度与沟通交流项目”,以促进创新药审评审批。
2016年,FDA共批准94个NDA(新药上市申请),包括24个优先审评以及70个标准审评,优先审评中位时间为8个月,比PDUFA V设定的6个月绩效目标要慢一些,标准审评已实现PDUFA V设定的10个月绩效目标。此外,FDA新收到共计1678个IND申请(新药临床试验申请),包括786个商业IND申请,892个研究IND申请。
CDER共立卷36项新分子实体(NME)药物申请,19个获批(批准率为61%),其中包括7个罕见病药物。NME优先审评的中位时间达到7.8个月,NME标准审评的中位时间达到12个月。FDA仍大力确保创新药能够进入加速的审评通道,通过优先分配审评资源、更快的沟通途径,从而保证加速这些具有潜在临床价值的创新药尽早上市。其中,68%的NME药物获得优先审评资格,32%的NME药物获得突破性治疗资格,37%的药物获得快速通道资格。同时,2016年批准的19个NME新药中,有37%是First-in-class新药,其中84%的新药首选美国市场并获得上市。
在仿制药审评审批方面,2016年,仿制药审评办公室(OGD)共批准了630个简略新药申请(ANDA),暂时批准183个ANDA(tentatively approved,指仿制药ANDA申请已获批,但由于专利权和/或独占权的原因无法在美国上市),预计在GDUFAⅠ到期前基本能实现GDUFA设定的审评绩效目标(90%)。
GDUFA的优先领域包括:1)仿制药上市后评价,通过开展患者仿制药替换原研药研究将患者观点更多融入到仿制药上市后评价中;2)开发复杂剂型和缓释药物的一致性评价方法,以推动各种类型产品的仿制药上市;3)吸入、眼科或胃肠道药物等局部用药的BE研究方法开发;4)治疗等效(TE)评价与标准建立;5)推进尖端计算和分析工具,以促进现代化的ANDA审评程序发展。
为促进更好推进等效、优质仿制药上市,OGD在2016年共发布了200多个具体产品仿制药开发指南,目前共有1500多个相关指南指导行业进行生物等效的仿制药开发。奥美沙坦酯(Benicar)、万艾可(Viagra)、可定(Crestor)、达菲(Tamiflu)等重磅炸弹药物也有首仿药获批。
OGD还通过与外部研究合作等方式,落实GDUFA的监管科学计划。包括新资助16位外部研究者开展监管科学研究活动,同时正在开展87项研究合作。
按照FDASIA要求,FDA于2017年3月29日发布《2015~2016 财年:监管科学进展报告》。该报告指出,FDA致力于改进临床和非临床评价工具以更好评估医药产品,加强内部IT工具以支持科学审评等监管活动。如在临床和非临床工具开发领域,FDA通过生物标记物资格认定程序批准了三个可用于临床试验设计的生物标记物,开发了体外方法工具更好支持仿制药生物等效性评估。
在儿科用药发展方面,《儿科最佳药物法案》(BPCA)与《儿科研究公平法案》(PREA)需要在2016年9月前向国会提交报告,并且之后每隔五年均需要将BPCA和PREA实施情况报告给国会。
FDA在完善儿科研究法律法规基础上,于2016年3月发布《儿科研究计划:首次儿科研究计划及修订的内容及程序》草案指南,指导行业如何递交首次儿科研究计划,进一步促进儿科药物的科学开发。
在创新审评途径方面,FDASIA建立的突破性疗法资格认定途径,授予了22个新药的突破性疗法。
2016年,FDA在生产场地注册、现场检查等方面引入了新的法规指南,通过全球合作促进检查工作的高效。FDASIA第712节中允许FDA与外国政府订立协议,互相认可对根据FD&CA注册的外国生产场地检查,以推动基于风险的场地检查实施。
近年来,FDA境外生产场地注册数量剧增。截至2016年,欧盟生产场地注册数量为1224个,较2011年增长了28%。FDA检查了392个欧盟生产场地(32%),然而仅有20个(5%)检查结果是官方行动(OAI),这种检查在一定程度上占用了FDA的检查资源。在这种背景下,2014年5月,FDA与EU正式签署了FDA-EU互认协议(MRI),此后FDA开展了13次针对欧盟药品检查部门的审计,计划2017年将开展更多审计工作。
对于FDA来说,2016~2017年是许多新法律政策酝酿诞生的时点。其中,PDUFA和GDUFA的再授权,以及21世纪治愈法案备受业界关注。
除加强产品安全性风险的控制、促进国际合作检查模式的开展外,FDA正着重发展以监管科学为核心的临床与非临床评价工具以及新证据开发,并且综合考虑到包括患者在内的多个利益相关方需求。
2016年是多个付费法案的第四个年头,许多付费法案将于2017年到期,需要FDA提交新的计划书提交国会重新授权。
其中,PDUFA和GDUFA作为FDA药品审评的两大主力法案,支撑着新药与仿制药之间的平衡。两大法案已广泛开展重授权前的讨论制定工作,并于2016年10月公布了承诺函。
处方药付费者法案(PDUFA)Ⅴ将于2017年9月到期,因此从2015年FDA已经开始研讨和制定PDUFAⅥ(2018~2022)。经过与利益相关者的6次谈判以及与行业的72次主题讨论,FDA最终于7月27日公布了PDUFAⅥ(2018~2022)绩效目标承诺函/绩效函(commitment letter/goals letter)。
作为PDUFAⅥ正式公开稿的风向标,该承诺函主要通过明确使用者付费资源的管理,设定审评绩效目标和其他程序优化目标,以促进公众获得安全、有效的创新药物。在2016年发布的这版承诺函中,几个重点项目包括:
在这个项目下,NME和原创性BLA这类创新程度最高的新药,将通过制定正式沟通计划继续顺畅PDUFAⅤ的原有沟通流程,实现申请人与FDA高度交互的沟通交流。
监管科学的发展涉及多个方面,包括:FDA和发起人沟通交流;突破性疗法资格认定使用;新替代终点使用的早期咨询;罕见病用药开发;组合产品监管;真实世界证据在监管决策中的使用;患者声音纳入到药物开发决策;监管决策的风险效益评价;推进从模型中获取信息的药物研发(MIDD);增强复杂创新的审评能力;增强用于研发与审评的分析数据标准的能力;促进生物标志物的资质认定途经(DDTQ)等。
这也是PUDFAⅥ经费重点分配的一个领域,通过采用新的科学方法,提高现有的识别、评价、预防、减少不良事件的工具,促进REMS数据收集进一步标准化,并融入医疗卫生体系中,提高上市前和上市后审评人员的沟通交流和协作,从而在整体上提高上市后安全信息的追踪和监管。使用者收费将会用于扩展前哨行动、整合药物警戒活动的上市后安全性评价、上市后安全性结果的及时、有效评估和沟通交流。
此外,PDUFAⅥ在首轮审评管理、药品专有名审评以减少用药差错、重大争议解决、临床试验暂停机制、特殊试验方案评价、审评与会议绩效目标等方面均设定了目标。
同PDUFAⅤ一样,GDUFA(仿制药付费者法案)Ⅱ也于2016年10月份发布承诺函,在GDUFA I基础上设定了新的绩效目标和项目框架。
标准审评时间为10个月,优先审评时间为8个月。符合条件的优先申请,如果审评人在ANDA提交两个月之前没有提供完整、正确的关于场地信息的预提交场地函,审评时间将延长至10个月。
对于2017年10月1日后批准的NCE产品,开发具体品种ANDA药物和证据生成的方法指南,以加速NCE产品仿制药上市进度,提高可及性。为了更好指导仿制药企业开展生物等效性试验,GDUFAⅡ中规定仿制药办公室(OGD)需要至少开发90%具体产品BA/BE指南,GDUFAⅡ正式出台后,这一绩效目标也将继续推动FDA建立生物等效性指南。
到2020年10月1日前,使用者可以电子访问非活性成分数据库。
原辅料DMF备案过程中,通过信息请求函(IR)和学科审评函(CRL)反馈给ANDA申请人,同时DMF持有人也在第一时间联动获知DMF审评情况。若DMF首轮审评存在缺陷,则通过DMF持有人通过电话会议解释阐述,若没有重大问题,将获得首轮通过函(First Adequate Letters)。
2018年10月1日前,FDA会发布Ⅱ型 API DMF批准后变更以及引用的ANDA的变更申请机制指南。
FDA将发布基于风险的场地选择模型;并与国外监管机构沟通合作,促进基于风险的场地检查;同时完善更新场地合规状态数据库。
2016年12月13日由奥巴马最终签署的《21世纪治愈法案》,是美国生物医药领域的又一重大立法,势必会对FDA一系列政策产生导向作用。
其中,第三章“开发”部分是该法案篇幅最大的一个章节,主要分为10部分(A~J部分),旨在对《联邦食品药品化妆品法案》(FD&CA)进行重点修订,并扩大了FDA的监管权力。其中很大篇幅在于促进新类型证据的使用,如适应性试验设计、贝叶斯统计学方法、生物标记物、替代终点等获得的数据,考虑特定患者的风险效益平衡。
法案第3001节明确规定FDA在药品审评时接受患者体验数据。患者体验数据包含患者在接受医疗诊疗和服务过程中的体验,以及患者对该疗法的偏好。患者体验数据可由患者及其家属、医护人员、研发人员和药品生产企业等进行收集。
为督促FDA实施该条,国会要求FDA在2021、2028、2031年6月1日前做出报告,反映FDA药品审批程序中涉及使用患者体验数据等工具的情况。
法案第3012节针对基因靶向药物或蛋白质变体靶向药物的发起人,允许使用该发起人之前获批的具有相同或相似技术的药物数据。
法案第3031节则提出,对于药品某些合格适应症的补充申请,FDA可仅审评数据摘要内容,但是申请人仍需附上所有相关数据。
另外,法案第3022节允许已批准上市药物增加新适应症,今后可通过提供真实世界证据获得批准,真实世界证据还可用于上市后研究要求(PSR)。
FDA今后将开展会议讨论,发布相关指南文件,支持发起人在新药申请中加入适应性设计和创新数据分析模型。
法案第3011节通过修订FD&CA,新增第507节,推动了FDA于2015年建立的药物开发工具资格认定程序(指南)明确立法,通过对生物标记物、动物模型、临床结局等药物开发工具在特定使用背景下的有效性进行认定,企业将无需提交经过验证的工具资料,从而缩短药物研发时间,减少药物研发的失败率。
目前,经获得资格认定的有6个生物标记物和2个临床结局指标。相信FDA会继续推动更多有价值生物标记物的开发与认定。
更好的生物标记物能更有效地评估未满足临床需求疾病的疗法;以患者报告结局(PRO)为安全性有效性证据,也是为了更好评价特定患者的疾病经历和疾病进展。增加NIH的投资将促进生物医学创新,有助于发现和验证更多有效的生物标记物等工具,而该款立法则有助于将有效的生物标记物等工具融入药物研发与审评过程中。
生物标记物的“发现”与促进药物的“开发”,两者缺一不可。如果基础研究不充分,将会导致未有效验证的生物标记物用作患者结局的验证,可能会将患者暴露在有风险且无效的疗法中。
每年至少200万美国人因耐抗生素感染致病,导致2.3万人死亡。对于许多感染,由于患者数量少,且缺乏参照疗法,传统的临床试验往往无法进行。有限数量患者抗菌药(LPAD)审批通道就是在这种背景下提出的。
法案第3042节规定,对于治疗威胁生命感染的抗菌药,FDA在批准上市时有一定灵活性,可基于有限数量患者数据批准抗菌药上市。同时,该抗菌药标签、广告上应带有“有限患者数据”标识等要求。
LPAD通道最初由总统议会科学技术咨询小组提议,在美国感染病协会以及1万多位医生和科学家的联合倡导下,旨在针对那些特定的、有限数量患者的细菌感染疾病,概念上与罕见病有点类似。在这种情况下,药品审评批准的风险效益平衡与普通药品不同,要考虑到患者未满足的临床需求,患者及时获得抗菌药的效益要与药品批准降低的风险平衡。
罕见儿科疾病优先审评券计划再授权,政策有效期延至2020年。更新罕见病用药资助计划,资助范围扩大到观察性研究,以促进研究罕见病的自然发展进程和采取治疗手段时的进展。
要求HHS部长协调人类受试者基本法规(45 CFR 46)和FD&CA下的受试者保护法规(21 CFR 46),进一步优化多中心的伦理委员会审评程序。取消医疗器械临床试验发起人只能使用本地IRB的规定,允许其使用中心伦理委员会对医疗器械临床试验开展伦理审评。
FDA今后有更大的灵活性,可豁免或修改最小风险临床试验的知情同意要求,HHS和NIH根据基本法规也可采取这种灵活性。对于治疗严重或危及生命疾病的研究新药的制药企业,应向公众公示其同情使用政策。
FDA今后可授予再生性治疗产品加速审批资格。
基于现有的医疗器械优先审评途径,在FD&CA中新增了医疗器械突破性疗法途径,该途径类似于药品的突破性疗法途径,旨在加快有早期临床效益医疗器械的研发和审评。
新增了扩大使用/同情使用条款,要求生产企业公示其同情使用政策,促进病危或亟需治疗患者参加临床试验。
对于促进组合产品的监管,要求:FDA与组合产品发起人会面,在研发早期就组合产品的最佳临床试验方案达成一致;说明监管组合产品的不同中心的争议解决程序应如何进行。
FDA将试点建立一个或多个跨中心部门,旨在发展和促进重大疾病领域疗法时药品(CDER)、生物制品(CBER)、医疗器械(CDRH)各大中心的工作协调。
2016年FDA对于药品相关法规的修订并不很频繁(见表1)。
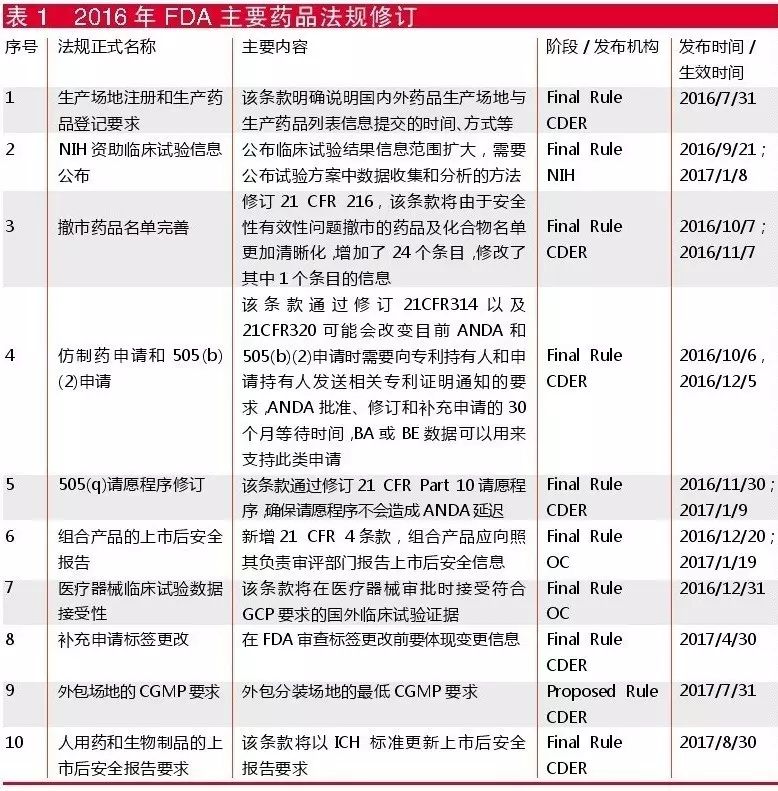
其中比较重要的一条是仿制药申请和505(b)(2)申请,该条款旨在更好落实2003年《联邦医疗保险处方药促进和现代化法案》的第十一章部分,该条款通过优化ANDA和505(b)(2)申请程序,进一步减少仿制药上市延迟以及带来的企业诉讼。此外,在美国,许多原研药生产企业会使用请愿程序影响ANDA和505(b)(2)批准过程,据FDA统计,这导致每年额外产生1700美元的行业成本和87,400美元的监管成本。因此FDA希望通过修订条款,减少不符合条件的505(q)请愿申请,从而减少行业和监管资源浪费。
FDA还通过新增条款,明确了组合产品的上市后安全报告(PMSR)要求,指出组合产品安全信息应向主要负责审评的部门汇报,从而减少重复报告,保证一致性和正确性。
另外,HHS发布临床试验透明度新规,收紧现有规定,要求研究者完全披露临床试验的成功与失败结果。
在指南发布方面,FDA于2016年共发布104个指南(包括50个最终指南和54个草案指南)。
其中,涉及临床方面的发布最多(见图1),如针对具体产品的临床试验设计与药物开发指南、抗菌药人体内药敏试验设计方法、生理药代动力学(PBPK)数据模式要求、临床试验种族数据的标准化收集等。
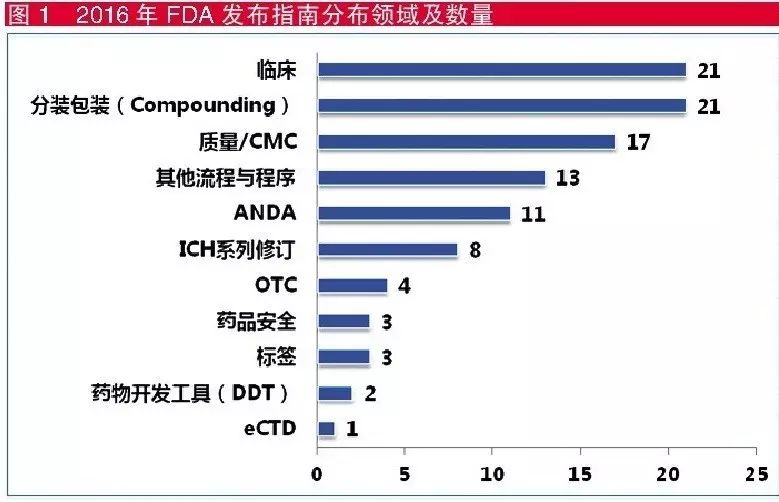
分装包装药品(coumpounding & repackaging pharmacueticals)方面也发布了21个指南,主要涉及分装包装厂地的无菌要求、分装要求等。
在ICH修订方面,E(Effficacy)部分E2C(R2)周期性风险效益评价报告第二版修订版、E11(R1)儿科药物临床研究第一版修订版、Q(Quality)部分Q7药物活性成分(API)的GMP指南,都已经修订后正式出台。另外,E17多中心临床试验设计基本原则、E18基因组数据采样和管理、S(Safety)部分S9抗癌药非临床评价Q&A、S3A毒性动力学的系统性评价指南,均是出台草案指南。
(本文作者来自沈阳药科大学国际食品药品政策与法律研究中心)
■编辑 余如瑾
★更多深度报道见《医药经济报》~ 










