高变异药物和高变异药物制剂分别指生物等效性试验中AUC 和Cmax的个体内变异系数≥30% 的药物和药物制剂。
个体内变异的来源包括影响生物利用度的生理学因素、药物的内在性质、物理化学性质、制剂因素及其他。
据统计,2003 -2005 年在FDA 做上市评价的仿制药制剂中,有约20% 为高变异药物制剂。进行高变异药物BE 试验时,需要更多的受试者才能达到足够的把握度。
但是,扩大受试者例数既会带来伦理学问题,也会增加申办者的经济负担。
因此,对于高变异药物的生物等效性评价问题,一直以来是各国研究人员和药政管理部门所关心的问题。
关于高变异药物的BE 研究,一些专家和机构提出的解决办法有:
① 增加样本量。
② 重复交叉设计或多剂量试验设计。
③ 放宽等效性判断的限值,这种方式又包括静态放宽、固定样本量放宽和比例标化的平均生物等效性。
比例标化的平均生物等效性方法是目前FDA 推荐的方法,即根据参比制剂的个体内变异程度放宽生物等效性判断的标准。
为了判断是否为高变异药物,需要采用重复交叉试验设计以评估药物的个体内
变异程度。
可采用部分重复的交叉试验设计( 3 周期) 或完全重复的交叉试验设计( 4 周期) ,其核心均强调参比制剂要重复给药1 次。
目前FDA 推荐比例标化的平均生物等效性方法,对于高变异药物,AUC 和
C
max
的等效性判断标准为: 上限/下限= exp [± ln( 1.25)·(
S
WR
/S
WO
) ]。
S
WR
表示参比制剂的个体内变异,
S
WO
是预先设定好的常数0. 25 ( 对应的个体内变异系数CV =25. 4%) 。
参比制剂个体内变异
S
WR
的界值定为0. 294( 对应的个体内变异系数CV = 30%) ,当
S
WR
≥0. 294 时,FDA 允许根据参比制剂的个体内变异程度放宽等效性判断接受的标准。
但是这种方法对受试制剂和参比制剂几何均值比( GMR) 的差别缺乏敏感性,有时会出现结果显示两个制剂是等效的,但两制剂GMR 的点估计值却落在80% ~125% 之外的情况。
而BE 试验模拟结果也表明,当个体内变异超过50% ~ 60%时,对GMR 的点估计值的限制则成为主导标准。
因此,FDA 还要求两制剂GMR的点估计值应在80% ~125%范围内。FDA 推荐的高变异药物的生物等效性评价具体算法见孕酮胶囊生物等效性的指导意见。
2010 年版EMA 指导原则采用的方法与FDA 相似,但也有一些不同。
EMA 只接受对
C
max
放宽接受标准,同时设定了最大的限度至69.84% ~ 143.19%( CV = 50%) ,对AUC 仍然采用80% ~125% 的标准。
对于高变异药物,EMA 的接受范围按下式计算: 上限/下限= exp(±k·
S
WR
) 。k 是一个为0.760 的常数,与比例标化的起点变异度( CV = 30%) 相对应,这样比例标化和非比例标化之间可平滑过度,以避免在个体变异略高于30%时,过度放宽标准。
而FDA 采用的比例标化常数有更大的自由度( 更宽的标准) 。FDA 和EMA 对置信区间的放宽标准如图1 所示。
图1 FDA( a) 和EMA( b) 生物等效性标准比较。
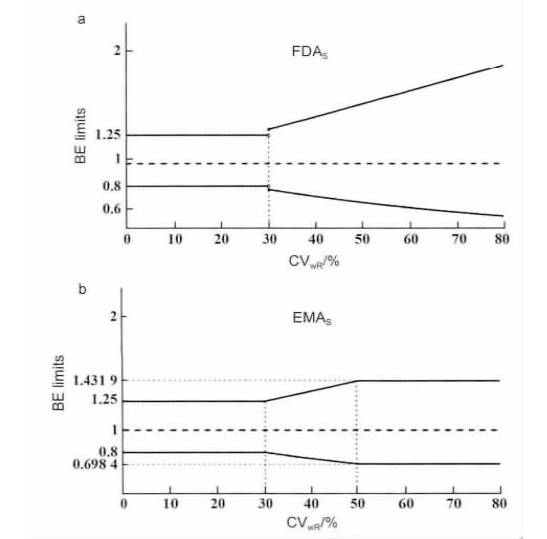
无论何种算法,采用比例标化的平均生物等效性方法,均可大大减少试验受试者的例数,并增加试验的把握度。
以阿仑膦酸钠为例,它属于高变异药物,AUC 和
C
max
的个体内变异约50%,1 项24 例受试者的双周期交叉试验把握度只有50%,尽管受试制剂和参比制剂参数的几何均值比为95%,其90%置信区间仍未落在80% ~125% 之间,要获得80%的把握度,至少需要106 例受试者。
如果采用3 周期重复交叉设计、比例标化的方法进行评价,31 例受试者可得到90% 的把握度。该方法已成功用于一项阿仑膦酸钠制剂的生物等效性研究。
EMA 对治疗指数窄的药物比较严格,AUC 的等效性标准缩窄至90% ~ 111. 11%,在
C
max
对安全性、药效或药物浓度监测特别重要的情况下,同样适用于上述范围。
FDA 在2012 年前对窄治疗指数药物的接受标准仍然是80% ~125%。而目前FDA 推荐的是比例标化的方法,在等效性限值90% ~111%基础上根据参比制剂的变异度放宽标准。
当参比制剂的个体内变异CV≤10% 时,等效性限值为90% ~111%; CV 在10% ~ 21%之间时,等效性限值比80% ~ 125% 窄; CV > 21% 时,等效性限值为80% ~125%。
另外要求受试制剂和参比制剂的个体内变异比值
S
WT
/
S
WR
≤2. 5。基于这一要求,只能采用完全重复的交叉试验设计( 4 周期) 。
FDA 推荐的窄治疗指数药物的生物等效性评价具体算法见华法林片生物等效性的指导意见。
EMA 的指导原则可以接受2 阶段试验设计,但要求必须在试验方案中预先规定,为减少I 类错误的发生,要规定调整后显著性水平。
在分析第1 阶段以及第1 和第2 阶段合并的数据时,使用94. 12%置信区间是可以接受的。EMA 和FDA 都对数据的剔除做了说明,如呕
吐和腹泻、非零基线浓度大
于
C
max
的5% 以及
AUC
0
~
t
覆盖
AUC
0
~
∞
少于80%的数据处理。
新版的FDA 指导原则指出: 对于半衰期较长的( 24 h 以上) 药物,如果药物分布和清除个体内变异较大,则不能截取部分AUC 来评价药物暴露量。
因为在给药后短时间内( 5 ~ 15 min) 未采集早期的样本,导致首个样本即为
C









