
肌肉松弛药及其拮抗剂的研究进展
闻大翔
*
,怀晓蓉
(上海交通大学医学院附属仁济医院麻醉科,上海200127)
[
摘要]
肌肉松弛药的临床应用为外科手术和机械通气创造了良好的条件,且提高了某些疾病的治疗效果。琥珀胆碱为当前临床上广泛应用的快速起效、超短时效的肌松药,但存在较多不良反应。因此,寻找、研发更为理想的肌松药以及肌松药拮抗剂是目前该领域的研究焦点。综述近年来肌松药及肌松药拮抗剂研究进展,对了解该领域的研究动态提供参考。
[
关键词]
肌肉松弛药;肌肉松弛药拮抗剂;药理作用
麻醉药理学在近几十年来取得较大发展,其中包括麻醉药物疗效和安全性的改善。多药物联合麻醉模式是现代麻醉实践的一个重要组成部分,近年来有较多研究关于镇痛和遗忘药物的改善,然而在半个世纪前就开始使用的肌肉松弛药(肌松药),在近
20
年里却罕有新的肌松药和肌松药拮抗剂面世,能够留在临床使用的肌松药则更少。理想的肌松药应具有起效快、副作用轻微、药物代谢对脏器依赖小、肌松作用能被肌松药拮抗剂快速安全拮抗等特点。尽管有明显的副作用,去极化肌松药琥珀胆碱仍然是目前临床上唯一使用的快速起效、超短时效的肌松药。截至目前由于药物起效时间慢和(或)各种副作用而限制了在研肌松药在临床的使用。为此,寻找、研发理想的新型肌松药和肌松药拮抗剂是目前该领域的研究焦点。本文对一些新型肌松药和肌松药拮抗剂的研究进展作一综述。
1
新型肌肉松弛药
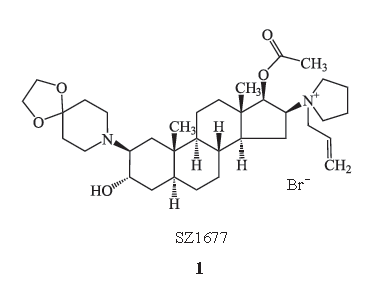
1.1SZ1677
1.1.1
化学结构式
SZ1677
(
1
)结构与罗库溴铵相似,它是
1-3[
α
-
羟基
-17
β
-
乙酰基
-2
β
-
(
1
,
4-
环氧乙烷
-8-azaspirodec-8-yl
)
-5
α
-
甾烷
-16
β
-il]-1-
(
2-
丙烯基)
4
氢化吡咯溴化物。
1.1.2
药理作用
Michalek-Sauberer
等
[6]
比较了
SZ1677ED
90
(
90%
的药物有效剂量,
25 μg· kg
–1
)和罗库溴铵
ED
90
(
100μg· kg
–1
)对豚鼠的喉肌和胫骨前肌的神经肌肉阻滞效力。结果显示,喉肌中两者起效相似,而胫骨前肌中
SZ1677
起效较慢,阻滞恢复较快;与现有其他肌松药相比,
SZ1677
起效快,维持时间较罗库溴铵短。研究提示,物种和肌肉种类的不同也是导致
SZ1677
神经肌肉阻滞过程不同的原因之一。
Vizi
等
在小鼠、大鼠、豚鼠和猫中,将
SZ1677
与现有肌松药对神经肌肉传导、
M2
和
M3
受体(
M
受体即毒蕈碱型受体)以及心血管系统等方面的影响进行比较。结果显示,
SZ1677
起效快,无蓄积,无明显不良反应,不引起血压下降,心动过速,不增加心脏交感神经释放去甲肾上腺素(
noradrenaline
,
NA
),
8
×
ED
90
(
90%
的药物有效剂量的
8
倍)无明显迷走神经阻滞效应,在产生良好神经肌肉阻滞效应和心血管不良反应之间有很大的安全剂量范围。与其他许多肌松药不同,
SZ1677
对人体心房组织没有类阿托品样效应,这与既往
Sato
等的研究结果相符。目前临床前研究表明,
SZ1677
比现有肌松药(如瑞库溴铵)在许多方面更具临床优势。
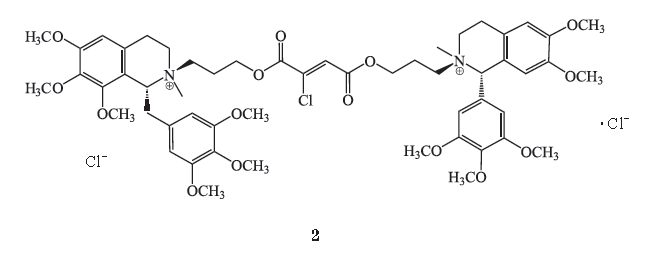
1.2 Gantacurium
1.2.1
化学结构式
Gantacurium
(更他氯铵,
AV430A
,
GW280430A
,
2
)是一个不对称四氢异喹啉氯延胡索酸盐。
1.2.2
药理作用
Gantacurium
是一种起效快、超短效的非去极化肌肉松弛药,属于双苄基异喹啉类肌松药,可用于气管插管和麻醉维持,在手术中维持骨骼肌的松弛,能被半胱氨酸拮抗。
1.2.2.1
临床前研究
在猴、犬、猫及豚鼠中针对
gantacurium
进行了多项临床前实验。这些研究显示,使用挥发性麻醉药麻醉时,
gantacurium
的
ED
95
为
0.06 ~0.1 mg · kg
–1
,
3
×
ED
95
剂量的作用时效为
12min
,几乎未观察到组胺释放和支气管痉挛。此外,超过
25
×
ED
95
剂量时可观察到组胺释放,但这个剂量远超过人体所需的插管剂量。在猫模型中的研究表明,
gantacurium
对于自主神经副作用有很高的安全比,相对于肌松药相关的溶血或神经节阻滞作用效力极低
[9]
。在豚鼠模型中的研究表明,
gantacurium
对于通过气道
M2
、
M3
受体作用介导的支气管收缩效力极低。
1.2.2.2
临床试验
Gantacurium
的临床试验始于
20
世纪
90
年代末,其研究结果总结于
2004
年首次发表
[12]
。使用丙泊酚
/
芬太尼
/N
2
O
麻醉的志愿者中,
gantacurium
的
ED
95
为
0.19mg · kg
–1
,起效时间为
2 min
,作用时效为
12 ~14 min
。研究表明,近
4
×
ED
95
剂量时
gantacurium
作用时效仅增加
5 min
, 恢复间隔[
5% ~95%
、
25%~75%
、
25% ~TOFr
(肌松恢复期)时
T4/T1
为
0.9
]
并没有改变
[12]
。以上研究还表明,与临床前研究相比,挥发性麻醉药增加了
gantacurium
效力,其所使用的
ED
95
倍数剂量的总量降低,接受
4
×
ED
95
剂量的志愿者血浆组胺浓度增加、血压平均降低了
17%
。比较
gantacurium
(
0.36
和
0.54mg · kg
–1
)和琥珀胆碱(
1 mg ·kg
–1
)对喉内收肌群神经肌肉阻滞的影响发现,
gantacurium
的起效时间分别为(
1.1 ±0.3
) 和(
0.9 ±0.2
)
min
,而琥珀胆碱(
1 mg ·kg
–1
) 为(
0.8 ±0.3
)
min
。比较
gantacurium
和琥珀胆碱在拇内收肌神经肌肉阻滞情况时发现,
0.36mg · kg
–1
gantacurium
的起效时间及
T1
恢复时间稍逊于琥珀胆碱,但
0.54mg · kg
–1
gantacurium
对拇内收肌神经肌肉阻滞效果几乎与琥珀胆碱(
1 mg ·kg
–1
)一样好。
在临床前实验和临床试验中,
gantacurium
的肌松效果持续时间很短,这很可能是由于其代谢不涉及肾脏或肝脏,而是通过半胱氨酸(非必须氨基酸)对其分子进行内收修饰,从而使药物分子与骨骼肌烟碱受体的亲和力降低来快速拮抗其神经肌肉阻滞作用。
1.2.3
不良反应
Gantacurium
的起效比琥珀胆碱慢,为获得满意的气管插管条件常会给予
3
×
ED
95
或更大剂量。在这一剂量范围,
gantacurium
可能引起明显的组胺释放,从而导致相应的副作用,例如可引起一过性的动脉血压下降,少数患者出现面色潮红。一般来说,血压下降
出现在诱导后
1 min
左右,与肌松药起效有关,且具有自限性(
2 min
),下降持续
30 s
后血压逐渐恢复。
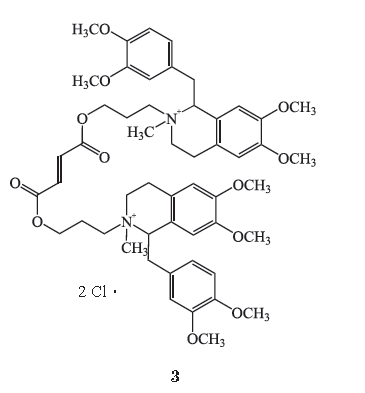
1.3CW002
1.3.1
化学结构式
CW002
(
AV002
,
3
)是近年研制的一种超短效非去极化肌肉松弛药
gantacurium
的化学结构类似物。
1.3.2
药理作用
与
gantacurium
相比,
CW002
能够被
L
型半胱氨酸水解得更慢,可用于气管插管和麻醉维持,在手术中维持骨骼肌的松弛。
1.3.2.1
临床前研究
与
gantacurium
一样,在多种动物模型中开展了
CW002
的临床前研究。与
gantacurium
相反,这些研究已证明不同物种间
CW002
的效价有所不同,在狗和兔子中其
ED
95
为
0.01mg ·kg
–1
,低于猫和猴(分别约为
0.03
和
0.04mg · kg
–1
)。尽管效价存在差异,但其相同倍数
ED
95
在不同物种间均产生中等作用时效。在狗模型中的实验表明,
3
×
ED
95
CW002
的起效时间为(
2.6±0.09
)
min
,作用时效为(
47±9
)
min
[13]
。虽然在低倍数
ED
95
时,血流动力学效应轻微,但剂量高达
25
×
ED
95
时,
CW002
轻度降低血压、心输出量和左心室收缩力。以上反应不直接归因于组胺释放,且体内实验中未观察到支气管痉挛现象。该研究也证实半胱氨酸能够在任何时间逆转
CW002
的神经肌肉阻滞效应。
随后在豚鼠和猫的研究中证实,
CW002
与
gantacurium
一样,几乎没有
M2
、
M3
受体介导的支气管痉挛作用。与对神经肌肉阻滞作用的效力相比,其对于溶血或神经节阻滞的效力很小。
1.3.2.2
临床试验
观察志愿者随
CW002
剂量增加(
0.02~0.14 mg · kg
–1
)的药效学
/
药代学变化的Ⅰ期临床研究已完成。在该研究中,使用丙泊酚麻醉诱导,且采用无肌松插管,采用
N
2
O
(
70%
)和七氟烷(呼气末
0.8%~1.2%
)进行麻醉维持,避免持续输注丙泊酚造成高血浆丙泊酚浓度,这可能会影响到
CW002
血浆浓度的测量。初步药效学
/
药动学数据显示,
CW002
的
ED
95
为
0.077mg· kg
–1
,(
1.5 ~2.0
)
×
ED
95
的作用时效为
55 min
,起效时间为
2.9min
,平均终末半衰期为
26 min
;未见血流动力学改变及组胺释放或支气管痉挛。
1.3.3
发展方向
CW002
发展成一个适合临床需要的肌松药还需进行更多动物实验及人体研究。有趣的是,
CW002
用于兽用麻醉受到较多关注,这很大程度上是因为目前一些动物可用的肌松药拮抗剂新斯的明(抗胆碱酯酶药物)的副作用显著,以及新型肌松药拮抗剂的研发成本较高。此外,还有些针对改善
gantacurium
维持效价、起效时间及保持短作用时效的同时减少组胺释放的研究正在进行中。近期初步活性结构研究显示,相较于
gantacurium
,另一个候选分子(
CW1759-50
)有类似的作用效果,而高剂量时其没有组胺释放相关副作用,这与其高自主神经安全性和非常低的呼吸道
M2
、
M3
受体效力相关。
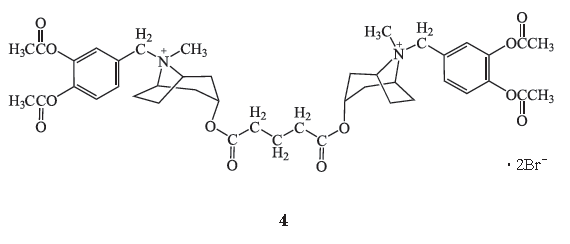
1.4 TAAC3
1.4.1
化学结构式
TAAC3
(
4
)是一种托品二酯衍生物。
1.4.2
药理作用
1.4.2.1
临床前研究
Gyermek
等
[21]
对鼠、兔、豚鼠、猫、猪、犬和猴进行了
TAAC3
与罗库溴铵在效价、起效速度、副作用等方面的比较研究,发现在不同种类动物中,与罗库溴铵相比,
TAAC3
的
ED
95
为
90 ~425 μg ·kg
–1
,起效时间(
0.8 ~1.0 min
)更快,恢复时间(
0.6 ~1.1min
)更短,持续时间(
1.8 ~3.5 min
)也更短;在猫和犬的实验中其快速起效甚至能和琥珀胆碱相媲美。研究者们认为,
TAAC3
快速起效不能用低效价进行解释,而与某些药物动力学因素有关。
1
×
ED
50
的
TAAC3
无心脏迷走神经阻滞作用,但是更大剂量能使鼠、豚鼠、猫特别是猪出现轻度心率加快、血压升高。另有报道称,(
5 ~ 10
)
×
ED
90
的
TAAC3
可使狗的血压明显下降,但与组胺释放无关。
1.4.2.2
临床研究
目前
TAAC3
已进入临床试验,但尚无详细研究结果报道。
2
新型肌松药拮抗剂
2.1
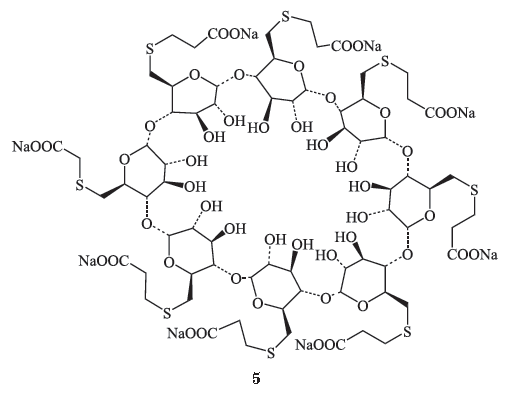
甾类肌松药特异性拮抗剂—— sugammadex
2.1.1
化学结构
Sugammadex
(
Bridion
,
Org25969
, 布瑞亭,
5
)是一种经修饰的
γ
-环糊精,具有亲脂内核心和亲水外端的圆柱体胶囊,其分子孔径及其结构上与罗库溴铵的疏水甾体分子骨架互补,以
1 : 1
的比例包裹外来分子如罗库溴铵形成宿主
-
外来分子螯合物,为无活性的紧密复合物。
2.1.2
药理作用
1
)
Sugammadex
影响甾类肌松药再分布,加速其与烟碱样乙酰胆碱受体分离,使游离肌松药分子浓度
急剧下降,直接消除肌松药的作用,从而拮抗神经肌肉阻滞。
Sugammadex
包裹外来肌松药分子如罗库溴铵形成的复合物主要分布在中央室(血浆)和细胞外液中,并以原形在尿液中排出。
2
)
Sugammadex
有选择性拮抗作用,可有效拮抗甾类肌松药,对非甾类肌松药和琥珀胆碱无拮抗作用。
3
)
Sugammadex
能高度选择性迅速消除罗库溴铵的不同深度肌松阻滞效应。给予罗库溴铵
0.6 mg · kg
–1
后,在
4
个成串刺激(
TOF
)恢复到第
2
个肌颤搐(
T2
)出现时,给予
2 mg · kg
–1
sugammadex
;重复给予罗库溴铵维持深肌松,当强直刺激后计数(
PTC
)
= 1
~
2
时给予
sugammadex
≥
4 mg · kg
–1
,
3 min
内神经肌肉传导功能就可以恢复;静注
1.2mg · kg
–1
罗库溴铵后,即刻给予
16 mg · kg
–1
sugammadex
,也能够立即逆转罗库溴铵的肌松作用。
4
) 与新斯的明相比,
sugammadex
对罗库溴铵的拮抗作用更有效、安全。
Carron
等的
meta
分析表明,在第
1
个肌颤搐(
T1
)抑制
90%
的阻滞下,
sugammadex
和新斯的明均具有拮抗作用,而
3 × ED
90
深度肌松阻滞时只有
sugammadex
发挥拮抗作用,而新斯的明即使给予很高剂量拮抗效果仍不明显;同时,
sugamaddex
拮抗罗库溴铵的术后不良事件(包括术后呼吸道相关并发症、心血管相关并发症)发生率明显少于新斯的明。
5
)在新生儿中的应用:
sugammadex
的相对分子质量为
2 178
,一般考虑其不能通过血脑屏障。既往研究表明,挥发性麻醉剂可影响血脑屏障的开放和完整性。
Satomoto
等
在啮齿类动物中的研究表明,暴露于
2
%七氟烷
6 h
后导致新生小鼠海马的血脑屏障超微结构异常。当
sugammadex
与七氟烷共同给予新生小鼠时出现脑中神经细胞凋亡显著增加(
150
%)。因此,在新生儿麻醉中,
sugammadex
与七氟烷联用可能会引起神经毒性,而单独使用
sugammadex
不诱导凋亡。在
2~12
个月的婴儿神经外科手术中应用
sugammadex
拮抗罗库溴铵诱导的神经肌肉阻滞的研究显示,在插管后使用七氟烷维持麻醉,
sugammadex
可迅速完全拮抗神经肌肉阻滞作用,且未见残余肌松作用和复发现象。在儿童中应用
sugammadex
拮抗神经肌肉阻滞作用仍需更多前瞻性和长久的后续研究。
6
)其他:有
sugammadex
成功应用于重症肌无力患者前列腺手术以及孕妇剖宫产全麻病例研究报道。而
40
名长达
10
年以上的吸烟者应用
sugammadex
拮抗罗库溴铵神经肌肉阻滞作用时,其到达每个
TOF
的时间相对于非吸烟者更长,但没有统计学意义。还需更大的人群样本量来确定吸烟是否影响
sugammadex
的使用效果。
2.1.3
不良反应和注意事项
1
)
2008
年
sugammadex
在欧洲获批上市后,已陆续在
70
多个国家上市。之后就有不少关于
sugammadex
不良反应的报道。
Sugammadex
最常见的不良反应是呕吐、口干、心动过速、眩晕和低血压。有报道称,
sugammadex
可能引起心脏
QT
间期延长。但其他随机、双盲、对照研究表明两者之间并无明显关系。
Sugammadex
的另一个副作用是严重的心动过缓。因此,
FDA
建议应密切监测患者的血流动力学变化,并及时处理。还发现一个不良反应是
sugammadex
与负压性肺水肿的发展相关,这可能由于膈膜为克服咽部肌张力维持咽部开放而产生的吸气力量导致,尽管当时患者的
TOFr > 0.9
。此外,
Palanca
等
的研究表明,
sugammadex
对于老鼠初级神经细胞培养具有毒性作用,并观察到
sugammadex
诱导的线粒体相关细胞凋亡的激活,与体内神经元胆固醇平衡的改变有关。尽管研究者指出,
sugammadex
穿过血脑屏障较罕见(大鼠中
< 3%
),但可能会出现无意中鞘内使用
sugammadex
的严重后果。最近韩国的一项研究表明,变异性心绞痛患者在给予
sugammadex
后
2 min
内出现冠状动脉痉挛
[32]
。研究者提出低镁血症和
sugammadex
均可能引起冠状动脉痉挛,但根据给药的时机,在此案例中可能
sugammadex
是更主要的原因,其可能与组胺释放有关。还有
2
例
sugammadex
和罗库溴铵复合物引起严重过敏反应的病例报告。已报道的
sugammadex
使用后的副作用,并不一定真正与
sugammadex
相关,但由于其在临床使用越来越多,应持续关注
sugammadex
的副作用。
2
)根据不同肌松药阻滞程度确定
sugammadex
的给药剂量,当深度阻滞
PTC
为
1
~
2
时给予
sugammadex
的剂量不低于
4 mg · kg
–1
。
3
)应用
sugammadex
逆转罗库溴铵的作用后
,
应间隔
6 h
后再用
sugammadex
才能有效,或改用苄异喹林类药物,如顺阿曲库铵。
综上所述,由于
sugammadex
对甾类肌松药的快速有效的拮抗作用,使肌松药在临床应用中更易满足围术期的外科开放或腹腔镜手术对深肌松的需要,同时又可满足术后快速麻醉复苏,降低肌松残余作用的风险性,从而减少患者术后复苏停留时间并加快出院,
sugammadex
的临床应用使得日间手术室多种短小微创手术得以实施。此外,
sugammadex
也适用于临床上使用甾类肌松药后“不能通气,不能插管”的紧急状况,增加患者的安全性。
2.2
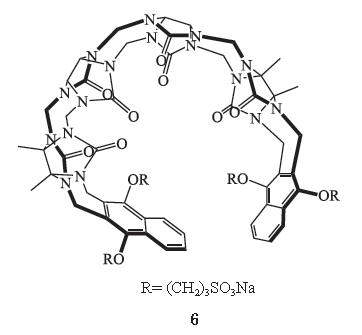
甾类和苄异喹啉类肌松药拮抗剂——calabadion
2.2.1
化学结构式
Calabadion 1
和
calabadion 2
(
6
) 属于分子容器(
molecular containers
)即葫芦脲(
cucurbituril
,
CB
)家族的非环状成员。
2.2.2
药理作用
2013
年美国麻省总医院
Hoffmann
等报道其成功研制对甾类和苄异喹啉类肌肉松弛药均有拮抗作用的葫芦脲家族无环化合物
calabadion
;该化合物既往被用作药物递送的载体,目前已证实其能覆盖苄异喹啉类和甾类肌松药的季铵位点,以阻止其与神经肌肉胆碱受体的结合。
sugammadex
能够在任何时间快速逆转而不需要抗毒蕈碱样药物,而
calabadion1
的作用机制似乎较特别,其能改变肌松药对胆碱受体的亲和力,而不是包裹肌松药并让其远离运动终板。
Calabadion2
同样能够拮抗苄异喹啉类和甾类肌松药的神经肌肉阻滞作用,对顺阿曲库铵的亲和力约是
calabadion1
的
5
倍。
有研究显示,
60
只大鼠静注罗库溴铵
3.5 mg· kg
–1
后静注
calabadion1
(
90 mg· kg
–1
),
(15 ±8) s
自主呼吸恢复,
(84 ±33s) TOFr
恢复到
0.9
;而静注顺阿曲库铵
0.6mg· kg
–1
后静注
calabadion1
(
150 mg· kg
–1
),
(47 ±13) s
自主呼吸恢复,
(87 ±16) s TOFr
恢复到
0.9
;均快于自发复苏和静注新斯的明和格隆溴铵,且静注
calabadion1
后不引起心率、心律、血压和血液
pH
的改变。
大鼠的体外和体内研究表明,
calabadion 2










