问题:
怎样才能让我的工作学习更有效率呢??
答案:
只需要点击图片上边蓝字
一药一世界
即可!
随着制剂多样性的发展和成熟,药用辅料在药品中的作用也早已从简单的赋形剂和附加剂发展到具有各种功能作用的重要组成部分。可以说,没有适宜、优质的药用辅料,就不会有质量和疗效可靠的制剂的开发和日常生产,这在正在进行的仿制药质量和疗效一致性评价和药品的创新发展过程中都已得到了验证。
现在,国家正在大力倡导药品的创新发展,改革力度之大史无前例。 在这里,也包括了药品的重要组成部分——药用辅料。犹如制药行业,中国的辅料行业也以仿制为主,质量和功能特性上和原研辅料也存在一定的差距。显然,辅料行业不太合适通过国家强制力推的仿制药一致性评价的方式对辅料进行质量和功能一致性评价。中国辅料行业的发展需要各方共同的推动,政策和制度的引导从而让资本、现有辅料企业及其它行业更有商务驱动进入和发展辅料行业将是非常重要的一部分。
在中国药品中广泛使用且被行业认可的优质辅料通常来自于美国、欧洲或日本。这些辅料的生产企业通常都是大化工企业,如陶氏化学、巴斯夫等。和欧美恰恰相反,中国目前进入药用辅料领域的化工企业却是少数。大化工企业对辅料产品的生产通常始于源头,更熟悉并容易控制辅料产品的质量和性能特点,基于用途、质量控制要求等的不同设定了不同的级别。在欧美,虽然药用辅料占大化工企业的营业额都是小比例(通常不到10%),但大化工企业还是愿意并相对容易进入药用辅料领域,这和欧美的药用辅料监管模式不无关系。以下为欧美药用辅料监管模式的一些介绍和思考供大家参考。
1. 欧美药用辅料的监管方式汇总如下表:
|
美国
|
欧盟
|
|
基于生产商的批准模式
|
否
|
否
|
|
辅料DMF系统
|
是(非强制要求)
|
否
|
|
使用先例原则
|
是(有公开的非活性物质数据库-IID)
|
是(无公开的数据库)
|
|
药典符合性要求
|
是
|
是
|
|
辅料工厂在监管部门的登记要求
|
否
|
个别国家有要求(如法国,但非注册审批)
|
|
总体上对药品注册进行技术要求
|
是(如元素杂质等要求)
|
是(如元素杂质等要求)
|
|
总体上要求药品企业对辅料供应商实施风险评估-GMP要求
|
是
|
是
|
根据上表,欧美药用辅料的监管模式进一步描述如下:
-
美国和欧盟均未对药用辅料实施审批制度,更未对药用辅料基于生产企业进行审批。
-
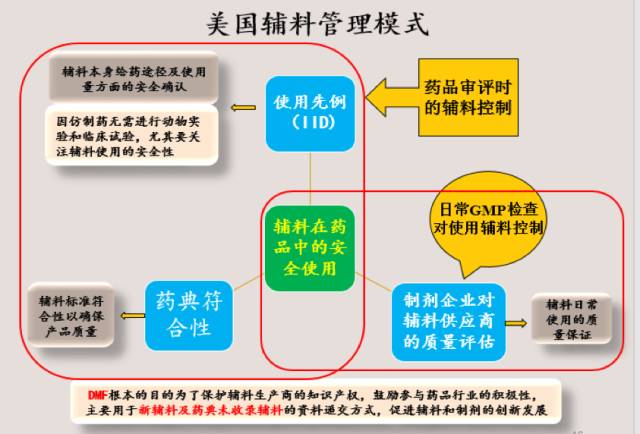
美国对辅料实施了DMF制度,但不是强制要求,辅料企业可以递交辅料产品的DMF,也可以不用递交辅料产品的DMF。尤其是美国药典收载的辅料,FDA更不建议辅料企业递交该辅料产品的DMF,有些辅料企业在辅料产品收载入美国药典后也主动不再维护已递交的该产品DMF。辅料DMF的申报与否,不会影响该辅料在药品中的使用。美国辅料DMF最主要的目的是在保护辅料生产企业机密信息的同时,允许其直接提供必要的信息至FDA以支持制药公司的药品注册。
-
欧盟并没有对辅料实施DMF制度。个别欧盟国家要求辅料生产企业在监管部门进行登记(但并非注册审批)。该类登记有利于监管部门掌握辅料企业在该国的现状,也便于需要时进行管理。
-
美国和欧盟均对药用辅料实施了使用先例原则,一个药用辅料如在药品中使用并进行了关联审评,也就意味着该药用辅料在特定的给药途径及使用量下进行了安全评估。该药用辅料如在别的相同给药途径的制剂中使用且使用量不超过其最大量,该药用辅料将不作为新辅料进行审评,审评所需资料也将显著减少。使用先例原则是基于药用辅料本身,而非具体辅料生产企业生产的辅料。如一个辅料没有在上市药品中的使用先例,即未在特定的给药途径下有使用历史或使用量超出了历史最大使用量,该辅料将需提供足够的安全性数据以支持在特定制剂中的使用。使用先例原则的要求确保了辅料本身在制剂中使用的安全性。
-
美国和欧盟均要求辅料在制剂中使用必须要符合相关药典要求,如在美国上市药品中使用的辅料需要符合美国药典,在欧盟需要符合欧洲药典。对进口制剂和本地制剂均采用同一标准要求。药典标准符合性的要求确保了辅料在制剂中日常使用的最低标准要求。
-
欧美监管机构通过总体上对药品注册进行技术要求间接对辅料进行了相关的要求,如动物来源的相关要求、制剂元素杂质的总体要求等等。
-
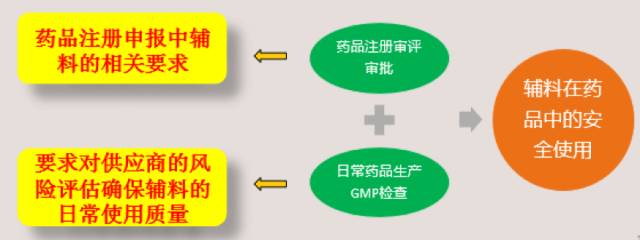
欧美监管机构在日常监管中,主要通过要求制药企业对辅料供应商(整个供应链,包括生产商和经销商)进行风险评估从而确保日常使用的辅料的产品质量。如制药企业没有对辅料供应商实施风险评估或风险评估不够到位,监管机构将对制药企业提出缺陷。在这方面,欧盟的要求更加详细和明确。2011年,欧盟伪造药品指令(2011/62/EU)颁布实施,其中第46条款(f)中要求:药品上市许可持有人(MAHs)应确认其使用的药用辅料是遵循适当的良好生产规范(GMP)生产的。2015年3月19日,欧盟正式发布“人用药所用辅料的GMP水平确定用正式风险评估指南” 官方文件(OJ2015/C 95/02)。该指南要求药品上市许可持有人对每一种药用辅料的生产和供应环节需要进行风险评估和实施适当GMP标准,从而确保病人的安全,并自2016年3月21日起实施。欧美监管机构都通常不会对辅料生产企业直接进行GMP检查。
-
不管是美国还是欧盟,均通过对“药品注册审评审批”和“日常药品生产GMP检查”的双管齐下来确保辅料在药品中的安全使用。
3. 如下为美国药用辅料整体管理模式的汇总,通过各个环节的要求以保证药用辅料在药品中的安全使用: