

摘
要:目的
通过构建
3D-
定量构效关系(
quantitative structure-activity relationship
,
QSAR
)
模型,研究黄酮类化合物对细胞色素
P450
酶
1A1
(
cytochromeP450 1A1
,
CYP1A1
)的抑制活性。
方法
借助
TopomerCoMFA
方法开展黄酮类化合物结构
-CYP1A1
抑制活性构效关系研究;通过体外孵育体系对化合物
CYP1A1
抑制活性进行评价,开展预测模型验证和优化;通过分子对接技术探究黄酮类化合物抑制
CYP1A1
活性的作用机制。
结果
构建的
3D-QSAR
模型交叉验证相关系数(
q
2
)为
0.800
,非交互验证系数(
r
2
)为
0.9650
,外部验证的相关系数(
r
pred
2
)为
0.9567
,表现出良好的稳定性和预测能力。利用该模型预测
9
种黄酮类化合物的
CYP1A1
抑制活性,实验值与预测值的相关系数
r
2
为
0.832 8
。分子对接结果显示,黄酮类化合物能与
CYP1A1
空腔内的
THR497
、
ASN222
、
ASN255
、
SER116
和
ASP313
等残基形成氢键,影响该类化合物的
CYP1A1
抑制活性大小。
结论
构建的模型具有良好预测能力与稳定性,为设计高活性分子提供理论参考,为新型
CYP1A1
酶抑制剂的开发提供新的思路。
细胞色素
P450
酶
1A1
(
cytochrome P450 1A1
,
CYP1A1
)是人体内重要的细胞色素
P450
酶。
CYP1A1
结构中共含有
512
个氨基酸,相对分子质量为
5.816
×
10
4
,包含
12
个
α
螺旋和
3
个
β
折叠。其中,
α
螺旋部分形成了
6
个底物识别位点
[1]
,可选择性识别、结合和催化不同类型的化合物
[2]
。研究表明,
CYP1A1
的活性与急慢性炎症、肺损伤、动脉粥样硬化、肿瘤等多种疾病的发生、发展密切相关
[3]
。
CYP1A1
能够代谢和活化杂环芳胺、工业芳胺、多环芳烃类
[4-5]
等多种环境致癌和致突变物质,促进
DNA
突变并最终形成肿瘤
[6]
。
CYP1A1
介导的前致癌物活化与癌症的发生发展关系密切,抑制其催化功能是癌症化学预防的潜在靶点
[7-8]
。黄酮属于天然多酚类化合物,广泛分布于蔬菜、水果、草药中,至今已发现上千种不同类型的黄酮类化合物
[9]
。黄酮类化合物具有保护心血管、抗氧化、抗衰老、调血脂、调节免疫等作用
[10]
。黄酮类化合物可以抑制
CYP1A1
酶的活性
[11-14]
,降低
CYP1A1
介导的前致癌物活化,预防癌症
[15]
。然而,黄酮类化合物与
CYP1A1
抑制活性的构效关系少见报道。
定量构效关系(
quantitative structure-activityrelationship
,
QSAR
)是基于配体的计算机辅助药物设计的主要方法之一,通过对化合物结构特征和生物活性进行定量分析,建立合理的数学模型,广泛用于新化合物的活性预测、筛选及设计。
3D-QSAR
不仅能够考察药物分子不同的构型或构象对其活性及性质的影响,还能够反映受体与配体的相互作用信息
[16]
。
CoMFA
和
CoMSIA
是目前应用最广泛的三维定量构效方法
[17-18]
。本研究采用第二代
CoMFA
(
Topomer CoMFA
)
[19-20]
构建黄酮类化合物与
CYP1A1
抑制活性的
3D-QSAR
模型,为黄酮类化合物与
CYP1A1
抑制活性的构效关系提供参考,并为筛选
CYP1A1
抑制剂提供更加快速有效的方法。
1
材料
1.1
药品与试剂
7,4′-
二羟基黄酮(
7,4′-dihydroxyflavon
,批号
AB151809
,质量分数为
97%
)购自德国
ABCR
公司;
4′,5′-
二羟基黄酮(
4′,5′-dihydroxyflavone
,批号
L14161
,质量分数为
98%
)、
5,6-
二羟基黄酮(
5,6-dihydroxyflavone
,批号
H27039
,质量分数为
97%
)购自
Alfa Aesar
公司;
7,8-
二羟基黄酮(
7,8-dihydroxyflavone
,批号
D1916
,质量分数>
98%
)、
6-
甲氧基黄酮(
6-methoxyflavone
,批号
M1346
,质量分数>
99%
)购自
TCI
公司;异黄腐酚(
isoxanthohumol
,批号
JOT-11423
,质量分数≥
98%
)、芫花素(
5,4′-dihydroxy-methoxyflavone
,批号
JOT-10632
,质量分数≥
98%
)购自成都普菲德
生物技术有限公司;
5,6,7-
三甲氧基黄芩素(
5,6,7-trimethoxyflavone
,批号
B29309
,质量分数≥
97%
)购自上海源叶生物科技有限公司;石吊兰素(
lysionotin
,批号
MB4495
,质量分数≥
98%
)购自美仑生物。
7-
乙氧基试卤灵为课题组自制;葡萄糖
-6-
磷酸(
glucose6-phosphate
,
G-6-P
)购自美国
Sigma- Aldrich
公司;葡萄糖
-6-
磷酸脱氢酶(
glucose-6- phosphatedehydrogenase
,
G-6-PDH
)、
β-
烟酰胺腺嘌呤二核苷酸磷酸(
β-nicotinamide adenine dinucleotide phosphate
,
β-NADP
)、
MgCl
2
购自美仑生物;
PBS
缓冲液购自中国医药集团有限公司;
CYP1A1
酶(批号
8191004
)购自
Gentest
公司。
1.2
仪器
SpectraMaX M4
酶标仪(上海美谷分子仪器有限公司);
MS-100
型恒温混匀仪(杭州奥盛仪器有限公司)。
2
方法
2.1
数据集来源与能量优化
通过查阅相关文献,选取
50
种已报道的对
CYP1A1
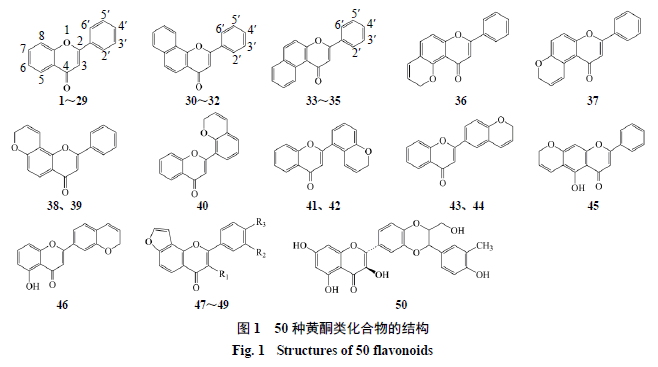
有抑制作用的黄酮类化合物,其化学结构与活性数据见图
1
和表
1
。
CYP1A1
抑制活性采用半数抑制浓度(
halfmaximal inhibitory concentration
,
IC
50
)表示,为更好地获得黄酮类化合物的结构与活性之间的线性拟合情况,将其负对数值
pIC
50
作为模型的因变量。
pIC
50
=
−lgIC
50
采用
Chemoffice16.0
构建化合物的三维结构,导入
SYBYL-X2.0
软件中,在
Compute Minimize
模块对化合物进行能量优化
[21-22]
,采用
Powell
能量
梯度法,选择
Tripos
分子力场,进行优化,
Gradient
设置为
21 J/mol
,分子电荷设置为
Gasteiger- Hunckel
,最大迭代次数设置为
1000
。优化完成后将其导入分子表单。
2.2 Topomer CoMFA
模型构建
Topomer CoMFA
方法是对传统
CoMFA
方法的一种改进,是
CoMFA
与
Topomer
技术的联合,能够在短时间内快速、准确预测化合物的活性并构建
3D-QSAR
模型。将
50
个黄酮类化合物按
4
∶
1
分成训练集和测试集。根据化合物取代基不同,分为不同类型的结构层,从不同结构层中随机挑选化合物组成测试集,剩下的化合物为训练集。以黄酮作为公共骨架,随机选择一个分子作为模板,切割分子的方式为“
Split into two
”,如图
2
所示,切出
2
个取代基(
R
1
、
R
2
)片段。软件自动识别剩余的化合物并以相同的方式进行切割,遇到程序不能自动识别的分子,选择与模板分子尽可能相同的方式手动切割,直至所有的训练集分子被切割完为止。

Topomer CoMFA
采用偏最小二乘法(
partial least squares method
,
PLS
)
[23]
建模分析,当所有训练集分子切割完成后,程序会自动计算每个分子片段的立体场和静电场性质,再以立体场和静电场性质描述符为自变量,以训练集化合物分子的
pIC
50
为因变量构建模型。测试集不参与
QSAR
模型的建立,仅用于模型的外部验证,以检测模型的实际预测能力。
2.3
实验验证
采用构建的
TopomerCoMFA
模型预测
9
种黄酮类化合物的
CYP1A1
抑制活性。
96
孔板中加入
100 mmol/L PBS
缓冲液(
pH 7.4
)及还原型烟酰胺腺嘌呤二核苷酸磷酸(
nicotinamideadeninedinucleotide phosphate
,
NADPH
)产生体系(
1 mmol/LNADP
+
、
10 mmol/L G-6-P
、
1 U/mL G-6-PDH
、
4 mmol/L MgCl
2
)和
CYP1A1
酶,总体积为
200 μL
,轻轻震荡混合。分别加入样品或纯乙腈,于恒温混匀仪
37
℃预孵育
3 min
;加入反应辅因子(
β-NADP
)起始反应;采用酶标仪连续监测反应
10 min
。产物试卤灵激发波长
560 nm
,发射波长
590 nm
。
2.4
分子对接
采用
SYBYL-X2.0
中的
Surflex-Dock
[24]
进行分子对接,对接所使用的蛋白酶晶体结构来源于
RCSB PDB
数据库(
http://www.rcsb.org/structure/4I8V
),晶体结构的
PDB ID
为
4I8V
[1]
,对接模式为
Surflex-Dock
(
SFXC
)。在对接前对蛋白进行处理,从复合物中提取配体
A/BHF602
,删除不需要的配体及所有水分子,给蛋白加氢加
Gasteiger-Hückel
电荷,设定距离配体分子
0.5 nm
范围内的所有氨基酸残基为活性口袋进行叠合,然后将模板分子
1
号化合物与蛋白晶体进行对接。用总打分函数、配体与受体对接的不适当程度和极性打分函数评价对接结果。总打分函数表示受体与配体的亲和能力,打分越高越好;不适当程度的绝对值越接近零,表示配体与受体对接时的不适当程度越小;当结合位点位于分子表面时,极性打分函数越高越好,当结合位点位于分子内部时,极性打分函数越低越好。
3
结果
3.1
Topomer CoMFA
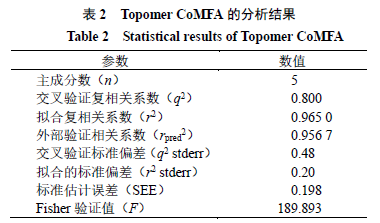
模型统计结果分析
对于所建
Topomer CoMFA
模型,交互验证复相关系数(
q
2
)、拟合复相关系数(
r
2
)和外部验证相关系数(
r
pred
2
)越大表示模型的相关性越好,预测能
力越强;
q
2
的标准误差(
q
2
stderr
)、
r
2
的标准误差(
r
2
stderr
)和标准估计偏差(
SEE
)越小证明模型质量越高。当
q
2
>
0.500
、
r
2
>
0.600
时,表明建立的模型具有显著的统计学意义
[25-26]
,当
r
pred
2
>
0.5
表明模型具有较强的外部预测能力
[27]
。本研究所建模型各参数如表
2
所示,模型
q
2
>
0.500
,
r
2
>
0.600
,表明模型具有较好的拟合与内部预测能力,模型
r
pred
2
=
0.9567
,表明模型具有较好的外部预测能力。
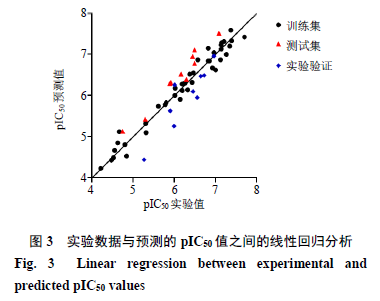
采用该模型对训练集和测试集化合物的活性进行预测,结果见表
1
。化合物训练集和测试集
pIC
50
实验值与预测值的线性回归图如图
3
所示,所有样本均匀分布在
45
°直线附近,表明所建模型具有良好的可靠性和预测能力。
3.2 3D-QSAR
分析
Topomer CoMFA
方法构建的
3D-QSAR
模型构建不仅可以获得模型评价参数,还可以获得各取代基的静电场和立体场三维等势图。其中,静电场等势图用红色和蓝色表示,立体场等势图用绿色和黄色表示。静电场中,红色代表在此区域引入吸电性基团有利于增大化合物分子的预测活性值,蓝色代
表在此区域引入供电性基团有利于增大化合物分子的预测活性值;立体场中,绿色代表在此区域引入大体积取代基有利于增大化合物分子的预测活性值,相反,黄色代表在此区域引入小体积取代基有利于增大化合物分子的预测活性值。
以化合物
1
(
pIC
50
=
6.01
)为模板生成的
TopomerCoMFA
模型的各基团静电场和和立体场三维等势图如图
4
所示。图
4-A
中,
R
1
分子片段的
C-3
附近有一小块黄色色块图,表明在此处引入小体积取代基有利于活性值升高。通过比较化合物
48
(
pIC
50
=
6.16
)、化合物
47
(
pIC
50
=
7.12
)的结构及活性值关系可以看出,当其他基团不变时,将化合物
48
R
1
分子片段
C-3
位上的羟基换成氢原子,此位置上取代基体积逐渐减小,化合物活性值随之升高。同时,
R
1
分子片段的
C-5
、
C-6
、
C-7
、
C-8
位在立体场中被大片绿色区域围绕,当其上的氢原子被更大体积的基团取代时,有利于
CYP1A1
抑制活性值的升高。如化合物
24
(
pIC
50
=
6.32
)、化合物
25
(
pIC
50
=
6.29
)与化合物
1
(
pIC
50
=
6.01
)相比,分别在
C-6
、
C-7
位用体积更大的氧丙炔基取代了氢原子,其
CYP1A1
抑制活性升高。图
4-C
中,
R
2
分子片段的
C-3′
、
C-4′
、
C-5′
位在立体场中被绿色区
域围绕,如化合物
21
(
pIC
50
=
7.70
)、化合物
22
(
pIC
50
=
6.19
)分别在化合物
1
(
pIC
50
=
6.01
)的基础上,在
C-4′
、
C-5′
位
用体积更大的氧丙炔基取代氢原子,活性显著提高。
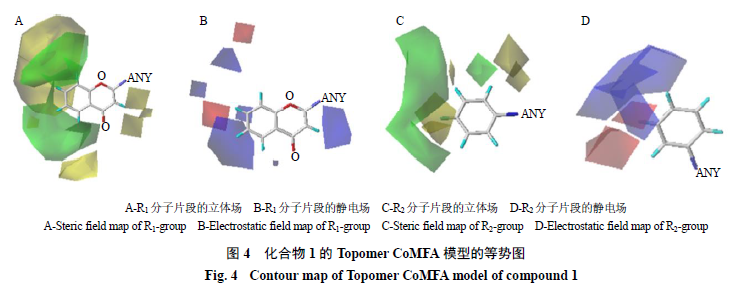
由图
4-B
可以看出,
R
1
分子的
C-3
、
C-6
位在静电场被蓝色区域所覆盖,当引入供电性基团时其
CYP1A1
抑制活性提高。对比化合物
6
(
pIC
50
=
5.89
)和化合物
11
(
pIC
50
=










