
药品试验数据保护是指原研药公司为获得新药 批准上市而向药品管理机构提交能证明该药品有效性和安全性的一系列试验数据。本文简析了药物试验数据保护制度起源、各国药物试验数据保护制度及我国药物试验数据保护制度
1984年,美国在Hatch-Waxman法案中首次引入药品试验数据保护制度,作为促进新药创新、平衡原研药企业与仿制药企业利益的一项重要举措。Hatch-Waxman法案,即《药品价格竞争和专利期补偿法》(Drug Price Competition and Patent Term Restoration Act)是 1984 年由美国国会批准通过,主要涉及加快和简化仿制药审批、原研药和仿制药的数据保护、专利期限延长和专利诉讼等主要内容。
1993年,WTO框架下最重要的知识产权协定《与贸易有关的知识产权协议》(TRIPS)第39.3款中,首次规定了WTO成员国对药品试验数据进行保护的义务,并确认药品试验数据保护是区别于专利、商标、著作权、商业秘密的一种新型的知识产权保护形式。
在TRIPs协议第39条第3款对试验数据保护(data protection)的规定是:当成员要求以提交未披露过的试验数据或其他数据,作为批准采用新化学成分的医药用或农用化工品上市的条件时,如果该数据的原创活动包含了相当的努力,则该成员应保护该数据,以防不正当的商业使用。同时,除非出于保护公众的需要,或除非已采取措施保证对该数据的保护,防止不正当的商业使用,成员均应保护该数据,以防其被泄露。
2015年10月,加拿大、日本、澳大利亚等多国签署《跨太平洋伙伴关系协议》(TPP),对药品试验数据规定了更高保护标准。
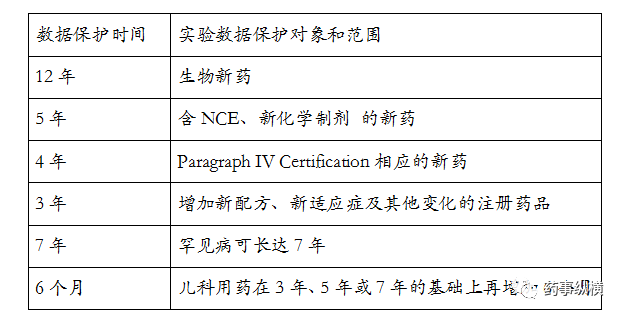
美国
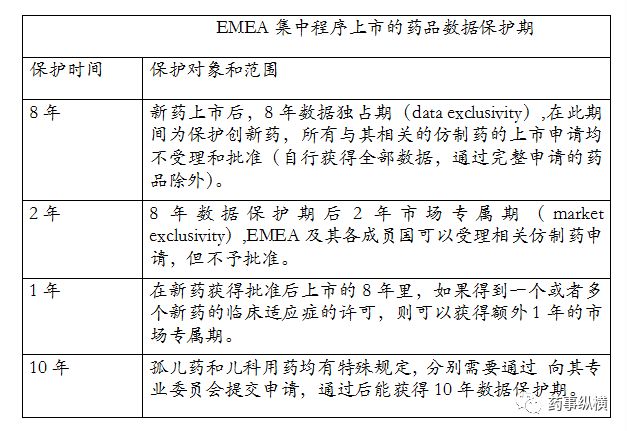
欧盟
欧盟国家药品上市的3个途径:
(1) EMEA集中程序
(2) 国家程序
(3) 互认程序

日本

加拿大

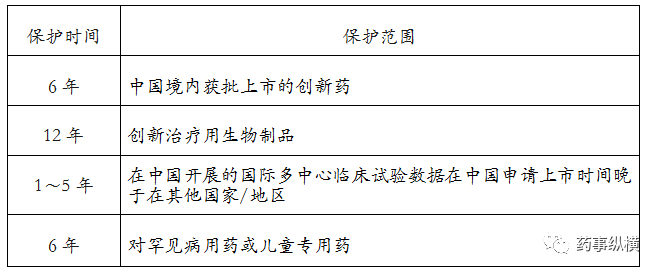
2018年4月25日,国家药品监督管理局组织起草并发布 了《药品试验数据保护实施办法(暂行)(征求意见稿)》。
实施主体
国家药品监督管理部门的药品审评机构负责药品试验数据保护的具体实施工作。
保护对象
(一)创新药;
(二)创新治疗用生物制品;
(三)罕见病治疗药品;
(四)儿童专用药;
(五)专利挑战成功的药品。
受保护的数据范围及条件
本办法中试验数据是指,药品上市申请人根据要求所提交的药品上市注册申请文件数据包中与药品有效性相关的非临床和临床试验数据,但是与药品安全性相关的数据除外,并应满足以下条件:
(一)以获得药品上市许可为目的提交药品注册申请资料中所要求提交的数据;
(二)提交药品注册申请前未公开披露;
(三)未依赖他人的试验数据或已公开发布的研究成果自行取得。
数据保护期限及范围
2018年7月10日,国家药品监督管理局组织制定并发布了《接受药品境外临床试验数据的技术指导原则》。
范围
本指导原则适用于指导药品在中华人民共和国境内申报注册时,接受申请人采用境外临床试验数据作为临床评价资料的工作。
接受境外临床试验数据的基本原则
申请人应确保境外临床试验数据真实性、完整性、准确性和可溯源性。
境外临床试验数据的产生过程,应符合人用药品注册技术国际协调会议(ICH)临床试验质量管理规范(GCP)的相关要求。
申请人应确保境外临床试验设计科学,临床试验质量管理体系符合要求,数据统计分析准确、完整。
接受境外临床试验数据的完整性要求
对于已有境外早期临床试验,后续在境内进行临床研发的,药品注册申请人应对早期临床试验数据进行评价,具备完整临床试验数据的,经与药审中心沟通交流后,可用于支持后续临床试验。
对于所有临床试验已在境外完成尚未上市的,应提供完整的境外临床试验数据包;已上市的,还应提供安全性、有效性更新数据,方可用于在中国的注册申请。
境外临床试验数据的提交情况及基本技术要求
对于境内外同步临床研发的,提交药品注册申请时,应按照《药品注册管理办法》的申报资料要求整理汇总境内外各类临床试验,形成完整的临床试验数据包,方可用于在中国的药品注册申请。
提交境外临床试验数据用于中国药品注册申请的资料,应包括生物药剂学、临床药理学、有效性和安全性资料数据。鼓励药品注册申请人采用通用技术文件格式(CTD)提交。
生物药剂学数据,应提供生物利用度和生物等效性相关的重要体外或体内数据和结果,为剂型确定和临床研发过程中制剂工艺优化提供支持依据和数据衔接。
临床药理学数据,主要包括药代动力学和药效学研究数据。药品注册申请人应从区域和人种等多角度进行种族敏感性分析,为境外临床试验数据适用于中国人群,及其有效性和安全性评价提供支持。
有效性数据,主要包括境外关键临床试验数据和在中国开展的临床试验数据,既要从整体上确证研究药物的有效性,还要分析中国亚组与总体人群的一致性。
安全性数据,包括境内外所有的用于安全性评价的数据,既要分析总体安全性,还要分析中国亚组与总体人群的一致性。
境外临床试验数据应支持有效性和安全性评价,药品注册申请人应考虑符合中国药品注册管理要求,在对完整临床试验数据包分析的基础上,对关键临床试验数据进行评价,以确证研究药物的有效性;遵循ICH关于接受国外临床资料的种族影响因素(E5)要求,分析中国亚组与总体人群的一致性,以支持境外临床试验结果外推至中国人群。
境外临床试验数据的可接受性
依据临床试验数据的质量,对临床试验数据接受分为完全接受、部分接受与不接受。
完全接受。境外临床试验数据真实可靠,符合 ICHGCP和药品注册检查要求;境外临床研究数据支持目标适应症的有效性和安全性评价;不存在影响有效性和安全性的种族敏感性因素。
部分接受。境外临床试验数据真实可靠,符合ICH GCP和药品注册检查要求;境外临床研究数据支持目标适应症的有效性和安全性评价,但存在影响有效性和/或安全性的种族敏感性因素。境外临床试验数据外推至中国人群的有效性和安全性评价存在较大的不确定性。药品注册申请人应根据影响因素分析情况,与药审中心进行沟通交流后,有针对性地开展相应临床试验。
不接受。境外临床试验数据在真实性、完整性、准确性和可溯源性方面存在重大问题,境外临床试验数据不能充分支持目标适应症的有效性和安全性评价,药品注册申请人应按照创新药研发思路,在中国开展系统临床试验,以支持在中国的药品注册申请。
对于用于危重疾病、罕见病、儿科且缺乏有效治疗手段的药品注册申请,经评估其境外临床试验数据属于“部分接受”情形的,可采用有条件接受临床试验数据方式,在药品上市后收集进一步的有效性和安全性数据用于评价。
药品数据独占权模式的药品数据保护制度,能够显著地弥补专利及其他保护形式的不足,推迟仿制药的上市,为原研药继续保持合法的市场垄断地位。对于某些由于种种原因无法得到专利保护的原研药来说,数据独占期的保护更加至关重要。
法律的功能就在于调节、调和以及调解多种错综复杂彼此冲突的利益,从而使其中大部分或最重要的利益得以满足,而同时使其他利益的牺牲最小,而药品数据保护制度就是平衡公共利益和新药投资者个人利益的法律保障。
参考文献
1.张文显.法理学[M].北京:高等教育出版社,2003,p370.
2.褚童.TRIPS协定下药品试验数据保护研究[M].北京:知识产权出版社.2015, p42-43.
3.《药品试验数据保护实施办法(暂行)(征求意见稿)》
4.《接受药品境外临床试验数据的技术指导原则》











