大家好,今天要和大家分享的是今年二月份发表在
Nat Commun(IF:12.121)
上的一篇PCAWG项目的文章,A deep learning system accurately classifies primary and metastatic cancers using passenger mutation patterns,文章从包含34种癌症在内的2778个样本的WGS数据(全基因组测序)中挑选了涵盖24种主要肿瘤类型的2436个肿瘤样本作为训练集,使用随机森林以及神经网络算法构建了分类器并在来源于不同WGS上游流程的数据进行验证。我们一起来学习一下吧
A deep learning system accurately classifies primary and metastatic cancers using passenger mutation patterns
深度学习系统根据乘客突变模式准确分类原发癌和转移癌
(分享者:科研菌-虾仁饭)
一、文章背景
在癌症中,肿瘤的起源器官和组织病理学是疾病自然史的唯一主要预测因素,包括预测肿瘤的生长速度,侵袭和转移以及总体预后。与此同时,研究表明基于肿瘤起源细胞的定点化疗比广谱化疗更加有效。但确定肿瘤起源这一项工作可能会在一些病例中遇到困难,在3% 的病例中,患者的肿瘤可表现为转移性肿瘤并且没有明显的原发肿瘤。在多重影像学研究不能确定原发癌的极端条件下,通常需要病理学家通过免疫组织化学染色辅助的组织学标准确定肿瘤的起源,但这一工作可能在部分低分化肿瘤不再表达免疫组织化学分型特异蛋白的干扰下变得非常复杂和耗时。
基于大规模的外显子组学研究表明,主要的肿瘤类型之间呈现出不同模式的体细胞突变。例如卵巢癌的突变特征为基因组重排率高,慢性粒细胞性白血病则通常含有t(9;22)易位导致的BCR-ABL基因融合等,最近的研究表明区域体细胞突变率和染色质可及性之间存在较强的相关性,并展现出从区域突变计数推断出起源细胞可能性。
这篇文章使用了包含34种癌症在内的2778个样本的WGS数据(全基因组测序),并使用同样的方法进行质量控制和体细胞突变分析,最终挑选了涵盖24种主要类型的2436个肿瘤样本作为训练集。
作者探究了是否可以通过机器学习技术,利用全基因组测序鉴定得到的体细胞突变模式来准确确定肿瘤起源器官和组织学。
二、文章思路
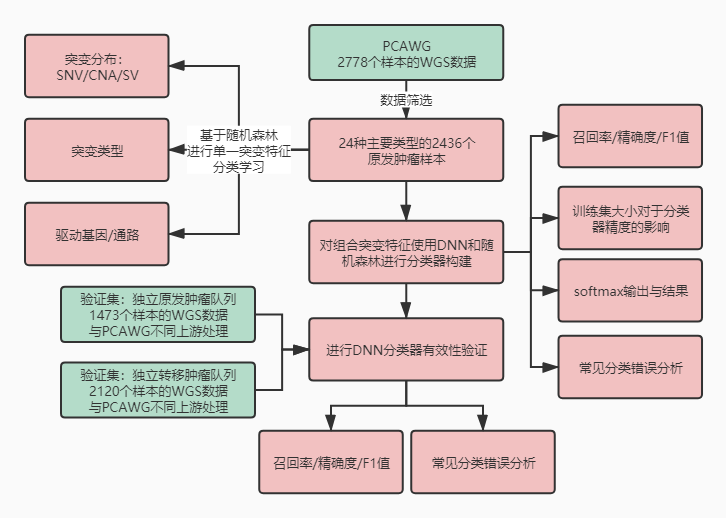
三、结果解读
1.数据筛选
PCAWG的全套数据包含来自于2778个供体的肿瘤,包括34种主要的组织病理学类型,并使用统一的数据处理流程。但由于PCAWG中的肿瘤类型分布不均匀且有部分肿瘤数量不足以充分训练并测试分类器。作者采用了以下的训练集数据筛选标准:
在此标准下,共获得涵盖24种主要类型的2436个肿瘤样本(表1)。
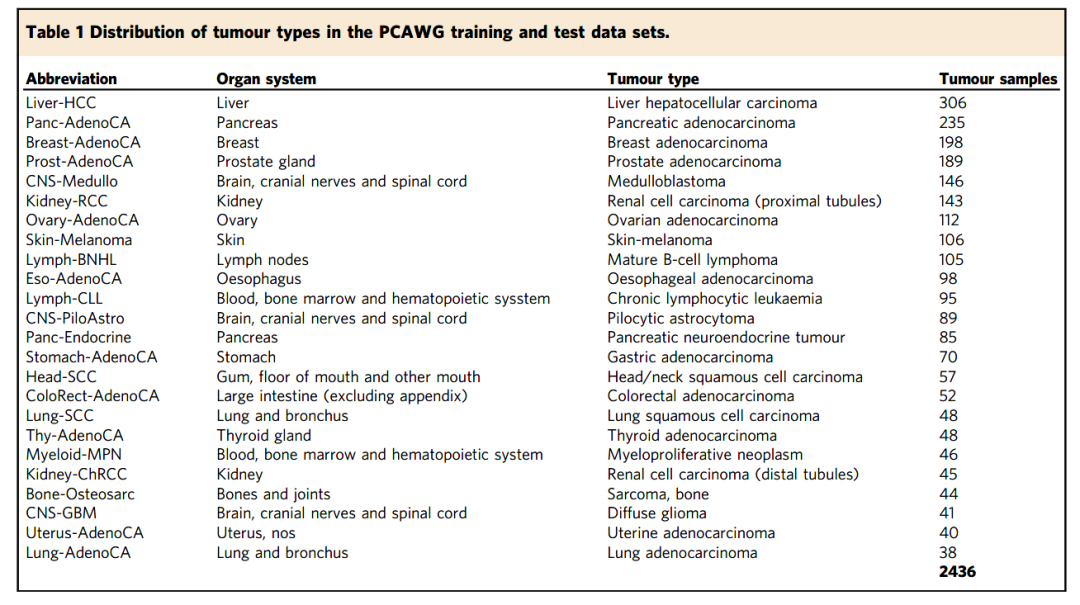
表1:PCAWG训练和测试数据集中的肿瘤类型分布
2.使用单一类别突变特征类型进行肿瘤分类
为了确定WGS测序中得到的不同突变特征对于肿瘤分类的价值,作者训练和评估了一系列基于肿瘤突变图谱(tumour mutation profile)中获得的单一类别变异特征作为输入数据的肿瘤类型分类器(表2)。
-
作者先对于每一个不同的变异特征类别进行随机森林分类器的构建,在训练时使用从每种肿瘤选择75%的样本作为训练集。
-
为了确定肿瘤最有可能属于的类型,作者将肿瘤突变谱应用于获得的24个肿瘤类型特异性分类器中,并选择其分类器得到结果最高概率的类型。
-
为了评估分类器的性能,作者使用四折交叉验证以获得准确性,召回率,精确度和F1评分。
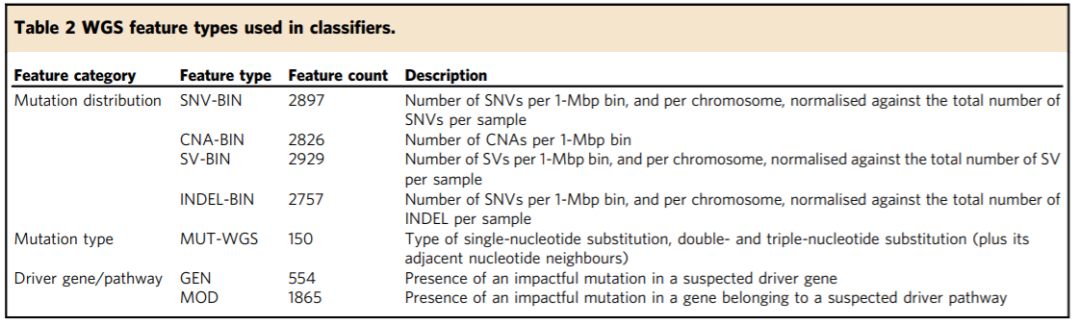
表2:构建分类器时使用的变异特征
在构建分类器时,作者对以下三个方面的突变进行了评估:
-
突变分布
-
由于癌症中体细胞突变率在基因组的不同区域存在较大的差异,并且在WGS中基因组序列的区域变异一个主要的共变因素是肿瘤细胞的表观遗传状态。作者将基因组分成不包含性染色体的3000个1-Mbp的区域,根据每个区域的体细胞突变数量创建样本特征。
-
突变率曲线(Mutation rate profiles)是针对体细胞单核苷酸变异(SNV),插入缺失,体细胞拷贝数变化(CNA)和其他结构变异(SV)独立创建的。这些突变大多数是非功能性的乘客突变。
-
突变类型
-
驱动基因/通路
-
一些肿瘤类型可以通过一些特定的改变来识别,如黑色素瘤的BRAF基因突变。
-
作者通过两种方法来鉴别:(1)基因是否受PCAWG癌症驱动因子工作组确定的驱动因子事件影响(2)是否在任何一个已知或可疑驱动因子途径的基因中存在一个有影响编码突变,这两点是根据先前PCAWG的工作确立的。
-
作者计算了影响蛋白质编码基因,lncRNA,miRNA的驱动因子突变,总共创建了约2000个与驱动通路相关的feature,这些feature描述了每种肿瘤的潜在基因和通路改变。
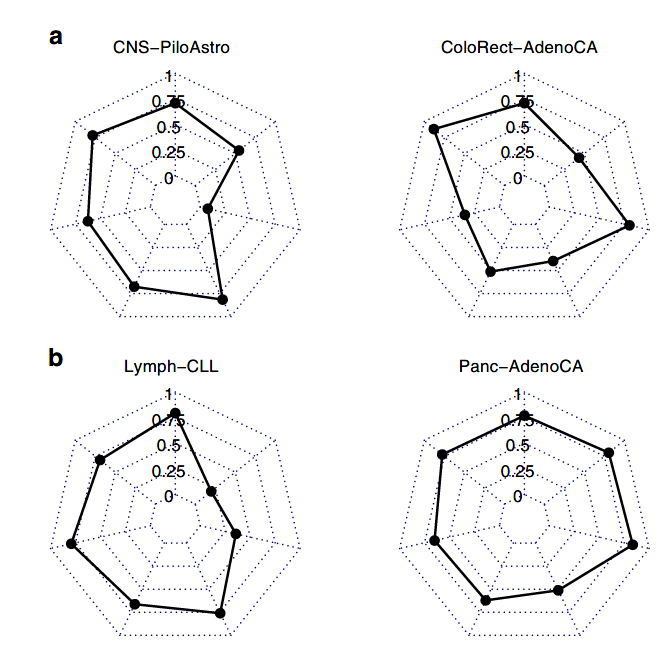
图1A/B:在不同肿瘤类型上7种单独特征类别的随机森林分类器的交叉验证衍生准确性(F1)得分
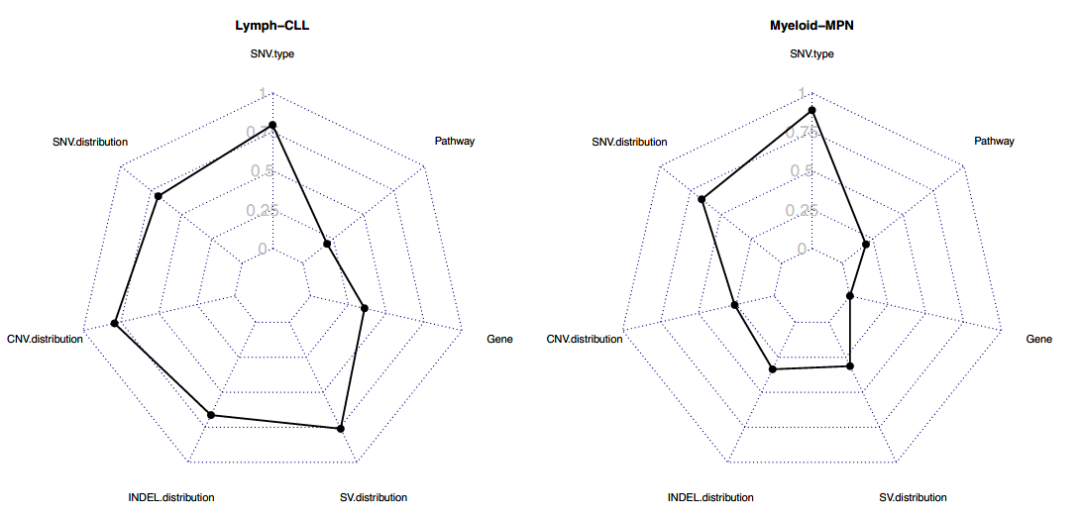
附图1(部分):在不同肿瘤类型上7种单独特征类别的随机森林分类器的交叉验证衍生准确性(F1)得分
通过单一类别变异特征构建的随机森林分类器准确度分布范围较大:
-
F1(召回率和精确度的谐波平均值)的中位数为0.42,范围为0.00至0.94。
-
九种肿瘤有至少一种性能较好的分类器(F1值=0.80)。
-
CNS-GBM, CNS-PiloAstro, Liver-HCC, Lymph-BNHL,Kidney-RCC, Myeloid-MPN, Panc-AdenoCA, Prost-AdenoCA and Skin-melanoma.
-
五种肿瘤的所有分类器均表现不佳(F1均小于0.6)
-
其他八种肿瘤的的F1值均在0.6-0.8之间。
在根据不同类别变异特征构建的分类器横向对比中:
-
与突变类型和分布相关的特征的分类器中观察到最高的准确性(图1B)。
-
根据驱动基因和通路的改变构建的分类器表现较差(中位F1值分别为0.33/0.27)。
-
SNV类型和分布构建的分类器中位F1值均为0.7。
3.基于突变特征类型组合的肿瘤分类
在根据单一类型的突变特征构建模型进行分类后,作者考虑是否可以通过组合两个或者更多特征来提高分类器的准确性,因此作者对于随机森林方法和基于多类深度学习/神经网络(multi-class Deep Learning/Neural Network, DNN)的模型进行了测试。
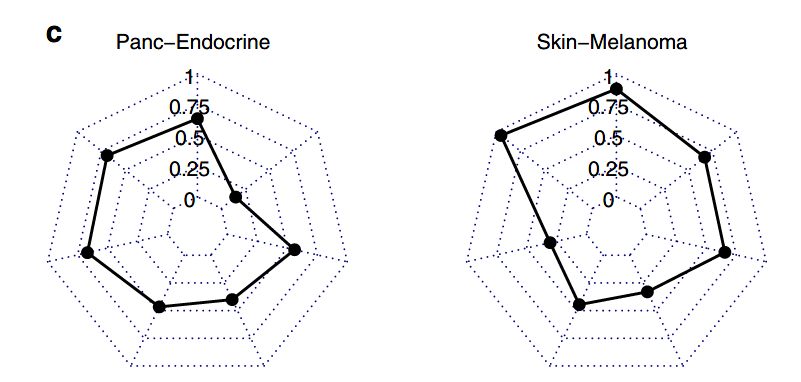
图1C:基于组合突变特征训练的分类器的交叉验证衍生准确性(F1)得分
-
在一系列特征类别组合中,基于DNN的模型总体上比基于RF的模型更为准确(DNN中位F1=0.90,RF中位F1=0.86,秩和检验p值<1.2e-7)(图1C)。
-
基于DNN的模型中,只考虑SNVs的拓扑分布和突变类型的模型总体准确性最高。增加驱动基因和通路改变的特征会稍微降低分类的准确性, 仅使用驱动基因和通路改变的特征进行训练会大大降低分类性能。
-
由于在在验证数据集中并不能统一获得CNV或SV的特征数据,所以作者没有在DNN中基于这些特征进行训练。
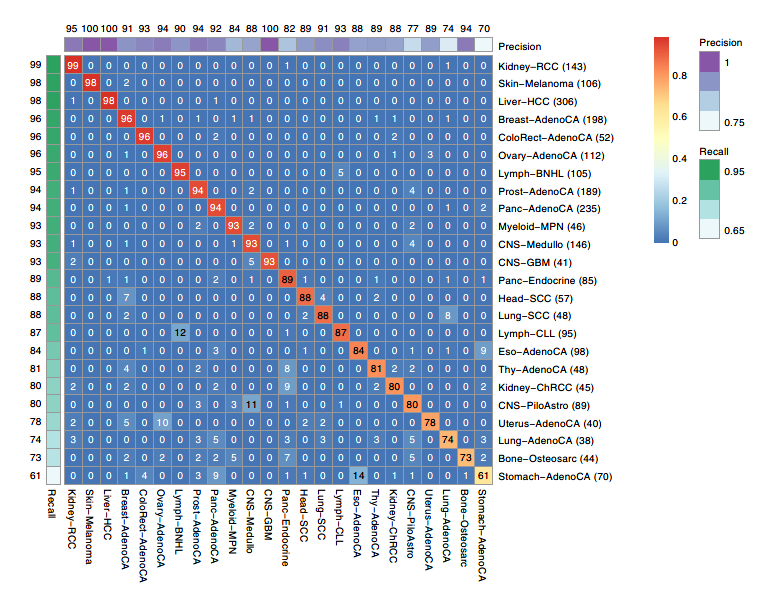
图2:DNN分类器的表现精度热图(行对应真实分类,列对应预测结果)
随后作者测试了10个独立训练的DNN分类器在PCAWG获得训练集的表现结果:
-
总体而言,24种肿瘤类型的分类准确率达到91%,但在个别肿瘤类型表现差异较大。
-
召回率(也称为灵敏度)范围从0.61(Stomach-AdenoCA)到0.99(Kidney-RCC)。
-
精密度(类似于特异性,但对数据集中的阳性数敏感)相当,比率范围为0.74(StomachAdenoCA)至1.00(CNS-GBM,皮肤黑素瘤和肝细胞癌)。
-
24种肿瘤中有21种的F1值大于0.80, 这21种中有8种在基于随机森林的分类器中的F1大于0.80。
-
表现最差的三种肿瘤是:CNS-PiloAstro(在10个DNN模型中的平均F1=0.79),Lung-AdenoCA(F1=0.77)和Stomach-AdenoCA(F1=0.67)。
由于PCAWG中不同种类样本的数量不同,作者研究了训练集大小对于分类器精度的影响:
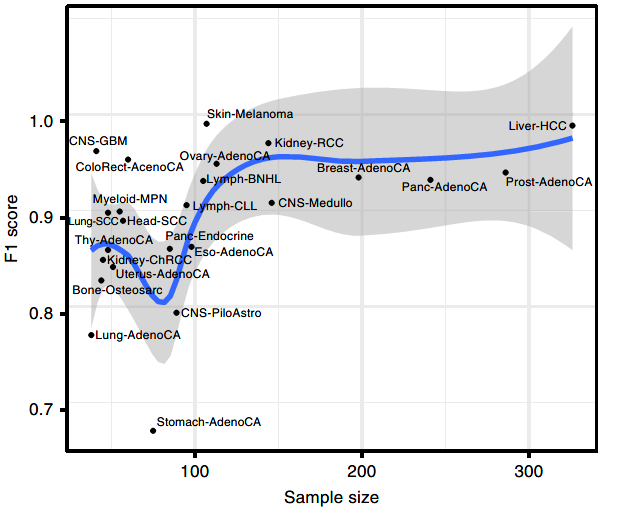
图3A:训练集大小和DNN预测精度的线性拟合
由于DNN模型的结果会进行softmax输出,并且通常选择概率最高的肿瘤类型作为分类器的最终选择。作者因此考虑了在根据概率排名前N个分类结果中包含正确类型的频率:
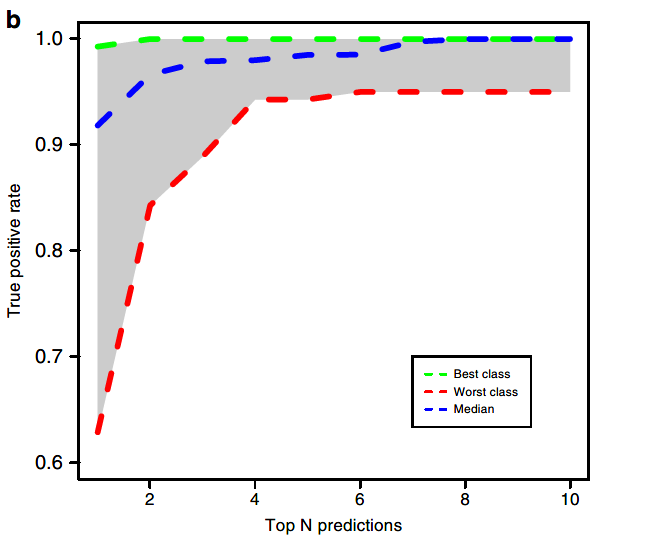
图3B:考虑根据概率排名前N个分类结果中包含正确类型的概率时分类器的准确性
4.常见的分类错误探究
作者对于基于DNN的模型中的分类错误进行了探究
作者推测胃癌和食管癌之间的高混淆率可能是两个位点之间相似的突变替代。有研究表明C-> A、C-> G碱基替换的一个子集常见于胃癌和食管癌。作者因此评估了单独使用突变分布而排除突变类型特征进行DNN训练的分类器效果。(附图2)
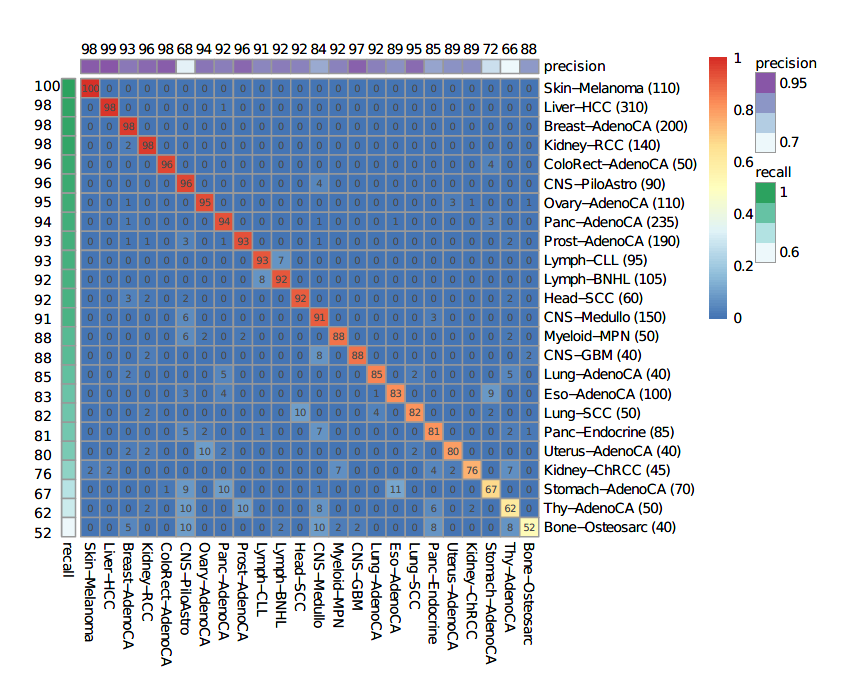
附图2:基于突变分布训练的DNN分类器的表现精度热图(行对应真实分类,列对应预测结果)
在其他常见的分类错误中,包括
而DNN对于不同起源细胞的癌症有着较好的分类效果:
5.对模型使用一组原发肿瘤进行验证
因为PCAWG的数据一个显著的特点是使用了一个统一的处理流程进行序列比对,质量控制等操作,而现实环境中用于训练分类器的数据集可能和用于测试的数据集在预处理时存在不同。
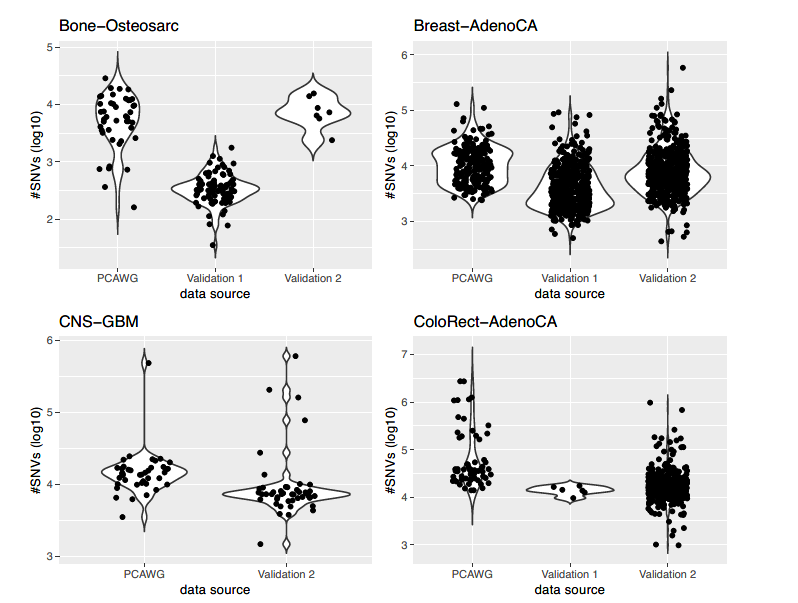
为了评估分类器在现实环境中的准确性,作者从非PCAWG项目中挑选了一系列肿瘤全基因组测序构建包含了1436个样本的独立验证集(附加数据4)。
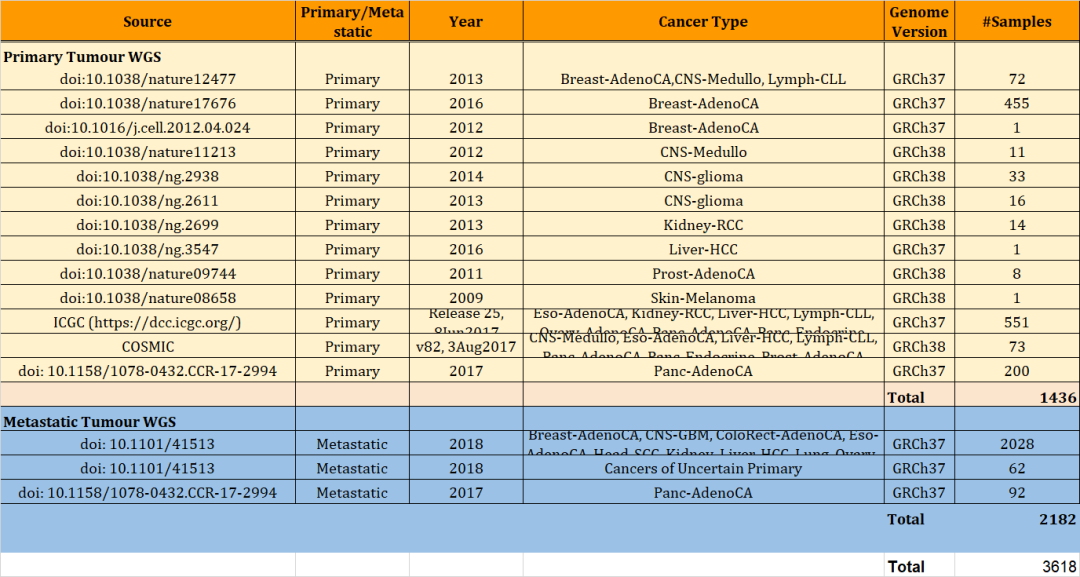
附加数据4:验证集详细信息
这个验证集包括14种不同的肿瘤,剩余的种类因为各种原因无法收集到足够数量合格的样本。部分样本的比对使用了GRCh37基因组。对除肝细胞外每个肿瘤队列进行突变负荷的比较发现没有显著差异(附图3)。