
雌激素在中枢神经系统疾病中的作用研究进展
任弋,季晖,唐苏苏,龚晓健
*
(中国药科大学药理学教研室,江苏 南京210009)
[
摘要]
雌激素不仅对生殖系统的生长发育具有重要的调节作用,而且参与中枢神经系统疾病的生理病理过程。近年来大量研究表明,其除了通过抗氧化应激、抗炎等途径抑制细胞凋亡外,还可通过促神经营养因子表达、改善多巴胺能神经元等发挥神经保护作用。综述雌激素在中枢神经系统常见疾病如脑缺血、阿尔茨海默病、帕金森病、多发性硬化症中的作用,为雌激素及其相关药物的研究提供理论依据。
[
关键词]
雌激素;脑缺血;阿尔茨海默病;帕金森病;多发性硬化症
中枢神经系统(
central nervous system, CNS
) 疾病如脑缺血、阿尔茨海默病(
Alzheimer, s disease
,
AD
)、帕金森病(
Parkinson, s disease
,
PD
)、多发性硬化症(
multiple sclerosis
,
MS
)等近年来发生率逐渐增高,但目前仍缺乏有效的对因治疗药物,因此寻找保护
CNS
结构及功能的有效药物至关重要。流行病学调查显示,这些
CNS
疾病发生率存在性别差异,且绝经后女性患病率远高于年轻女性。雌激素是人体内常见的一种甾体类固醇激素,包括雌酮、
17-
β
雌二醇(
17
β
E2
)及其代谢产物雌三醇。近年来研究表明,雌激素不仅在生殖系统的生长发育中有重要的调节作用,而且参与
CNS
疾病的生理病理过程。它除了依赖受体途径发挥作用外,还可通过抗氧化应激、抗炎等途径抑制细胞凋亡。因此,雌激素的神经保护作用是归因于不同神经生物学信号共同影响的结果。本文就雌激素在常见的
CNS
疾病如脑缺血、阿尔茨海默病、帕金森病、多发性硬化症中发挥的作用,予以综述。
1
雌激素在脑缺血中的作用
脑缺血是由大脑血流供应不足引起的
CNS
病变,具有高致残率及高病死率等特点。脑缺血的病理学机制复杂,脑组织血氧供应不足会导致氧自由基生成过量、细胞内钙超载、兴奋性氨基酸毒性作用、神经炎症等,造成局部神经元受损和神经学功能障碍。实验发现,雌激素具有缩小脑缺血模型动物脑梗死体积的作用,而这与雌激素的神经保护及血管保护作用密切相关。
研究发现,雌激素具有一定的抗氧化效应,可有效抑制活性氧自由基(
reactive oxygen species, ROS
)的过量生成,减少细胞凋亡的发生。体外研究发现,雌激素不仅能与高浓度的水溶性抗氧化剂及谷胱甘肽协同发挥抗氧化应激作用,还可通过增加线粒体抗氧化酶锰超氧化物歧化酶(
manganese superoxide dismutase,MnSOD
)的活力发挥抗氧化效应。同时,雌激素具有维持细胞内
Ca
2+
稳态的作用,谷氨酸主要位于大脑皮层和海马等部位,是
CNS
主要的兴奋性神经递质,在脑缺血的神经损伤过程中具有重要影响。脑缺血发生后谷氨酸病理性增多,表现出诱导神经元损伤的神经毒性作用,雌激素可通过丝裂原活化蛋白激酶(
mitogen-activated protein kinase, MAPK
)途径减少线粒体
Ca
2+
的内流,保护线粒体免受损伤,进而保护神经元。而在低浓度谷氨酸环境下,雌激素可通过增加神经元
Ca
2+
内流及调控
N
-
甲基
-D-
天冬氨酸(
N
-methyl-D-aspwhcacid, NMDA
)受体作用, 增加神经元对谷氨酸作用的敏感性,增强神经系统功能。
脑缺血损伤后,
CNS
免疫细胞小胶质细胞及星形胶质细胞被激活,炎症介质大量产生,触发神经系统炎症反应,持续的慢性炎症反应会导致神经功能受损,甚至更多神经元的死亡。研究发现,生理学浓度的
17
β
E2
具有显著的抗
CNS
炎症的作用,在青年及中年动物脑卒中模型中,
17
β
E2
可抑制促炎转录因子、核因子
κB
(
nuclear factor of kappa B, NFκB
)表达, 从而抑制许多细胞因子如肿瘤坏死因子
-α
(
tumor necrosis factor-α, TNFα
)、单核细胞趋化蛋白
-1
(
monocyte chemoattractant protein-1,MCP-1/CCL2
)、白细胞介素
-6
(
interleukin, IL-6 )
的转录。对小胶质细胞和星形胶质细胞,雌二醇可通过结合雌激素受体(
estrogen receptors
,
ER
)
2
种亚型
ERα
和
ERβ
而共同发挥抗炎效应,
ERα
可通过激活磷脂酰肌醇
-3-
羟激酶(
phosphatidyl inositol 3-kinase, PI3K
) 通路,增强
NFκB
活力并阻止其转运至胞核,
ERβ
对星形胶质细胞神经炎症具有调控作用,可通过上调神经珠蛋白,保护神经元免受缺氧、缺糖及氧化应激损伤。研究还发现,
17
β
E2
对白细胞
-
内皮细胞黏附作用的影响,是通过减少炎症细胞分子
e-
选择素(
endotheliumselectin,e-selectin
)、细胞间黏附分子(
intercellular celladhesion molecule-1, ICAM-1
)及血管细胞黏附分子
-1
(
vascular cell adhesion molecule-1, VCAM-1
)的信使核糖核酸(
messenger ribonucleic acid, mRNA
)表达所引起。
此外, 体内实验发现,
17
β
E2
对内皮素
-1
(
endothelin-1, ET-1
)诱导的脑缺血损伤具有保护作用,但在阻断胰岛素样生长因子(
insulin-like growth factor,IGF
)受体后,该保护作用消失,这表明
17
β
E2
对内皮素
-1
(
endothelin-1, ET-1
)诱导的脑缺血性损伤的保护作用依赖于
IGF
的作用而存在。越来越多的证据表明,雌激素可通过与
IGF
受体的相互作用发挥神经保护作用。
除神经保护作用外,近年研究显示,
17
β
E2
可通过基因组和非基因组机制增加脑内内皮型一氧化氮合酶(
endothelial nitric oxide synthase
,
eNOS
)表达,提高
NO
利用率从而促血管舒张,增加脑血流量并改善微循环,对脑缺血损伤具有保护作用。血脑屏障(
bloodbrain barrier, BBB
)是控制血液与脑组织之间物质交换的屏障,对
CNS
的稳定性具有重要作用。脑缺血后血脑屏障结构功能受损,
BBB
通透性的增加,导致白细胞浸润透过血脑屏障,局部炎症反应增多及持续性的神经凋亡增多。雌激素可通过
PI3K/
丝苏氨酸蛋白激酶(
serine-threonine kinase, Akt
)通路激活神经元细胞膜上葡萄糖转运体(
glucose transporter-4 protein, GLUT-4
),提高葡萄糖在脑内的摄取和转运,减轻由局部缺血及血管内皮生长因子诱导的大脑皮层
BBB
通透性改变。紧密连接蛋白存在于相邻血管内皮细胞间,对毛细血管的结构维持及血管内皮细胞膜的完整性具有重要意义,脑微血管内皮细胞的紧密连接是构成
BBB
的基础结构,雌二醇可通过调控紧密连接闭合蛋白和密封蛋白
-5
的
mRNA
及蛋白表达水平影响脑微脉管系统及血管内皮细胞功能,保护血脑屏障结构及功能。
2
雌激素在阿尔茨海默病中的作用
AD
是一种以记忆减退及认知功能障碍为主的神经退行性疾病,病理学特征主要为神经纤维缠结、老年斑和神经元突触的改变。其发病机制复杂,主要学说包括胆碱能神经元假说、
β-
淀粉样蛋白级联假说等。近年研究发现,雌激素具有调控胆碱能神经系统、抑制
tau
蛋白过度磷酸化、减少
β
淀粉样沉积、改善突触可塑性等作用,具有改善绝经后妇女
AD
的记忆认知功能的作用。
基底前脑内含有大量胆碱能神经元,可参与多种神经功能,对记忆认知功能具有重要意义,但其功能易受年龄影响,是
AD
的主要发病机制之一。雌激素可调控
CNS
的内源性神经递质乙酰胆碱(
acetylcholine,ACh
)水平,发挥神经保护甚至减轻认知功能障碍。胆碱乙酰转移酶(
choline acetyltransferase, ChAT
)是乙酰胆碱合成的关键酶,也是胆碱能神经元的标志酶,该酶活力的降低与年龄相关性的记忆受损有关。
17
β
E2
对
ChAT
的转录与翻译有促进作用,并能促进乙酰胆碱的合成和释放,从而改善机体的认知功能。早
年
Gibbs
等
研究发现,
17
β
E2
治疗可增强成熟去卵巢(
ovariectomized,OVX
)大鼠基底前脑不同亚区的
ChAT
蛋白活力并增加其
ChAT
免疫活性细胞数目,其后续研究表明,
17
β
E2
治疗不仅可改善记忆功能,还可通过基底前脑胆碱能系统改善非病灶性基底前脑损伤动物的认知障碍。除采用
17
β
E2
之外,
Acosta
等对中年
OVX
大鼠采用结合型雌激素治疗(
conjugatedequine estrogens, CEE
),发现基底前脑
ChAT-
免疫活性神经元数量增加,乙酰胆碱的代谢紊乱得以平衡,动物的空间记忆能力明显改善。近年研究发现,
AD
患者胆碱能神经元的退变与神经营养因子的缺乏具有密切关系,而雌激素与神经营养因子在部分脑区可相互影响,
ER
和神经营养因子具有相互影响调节的作用。动物体内实验表明,雌激素治疗可增加基底前脑、额叶皮层及海马中的神经营养因子及其受体
mRNA
表达水平,升高认知相关脑区的神经生长因子(
nervegrowthfactor, NGF
) 和脑源性神经营养因子(
brain-derivedneurotrophicfactor, BDNF
)表达水平,对大鼠
AD
模型具有显著的神经保护作用。
脑内
tau
蛋白过度磷酸化,造成神经元纤维缠结,是
AD
的主要病理学变化。研究发现,雌激素对
tau
蛋白磷酸化相关的蛋白激酶和磷酸酶活性具有调控作用,而这与对糖原合成酶激酶(
glycogen synthasekinase, GSK
)通路的调控作用有关。雌激素具有降低
GSK-3β
活性的作用,黄体酮则具有减少
GSK-3β
和
tau
蛋白表达的作用,均可减少
tau
蛋白磷酸化。此外,雌激素可通过激活细胞外因子
/β-
连环蛋白(
Wnt/β-catenin
)信号通路,抑制
c-Jun
氨基末端活化蛋白激酶(
c-Jun N-terminalprotein kinase, JNK
)、
Wnt
信号通路抑制因子(
dickkopf-related protein 1, Dkk1
)及蛋白激酶
A
(
proteinkinase A,PKA
)活性,减少
tau
蛋白的磷酸化。
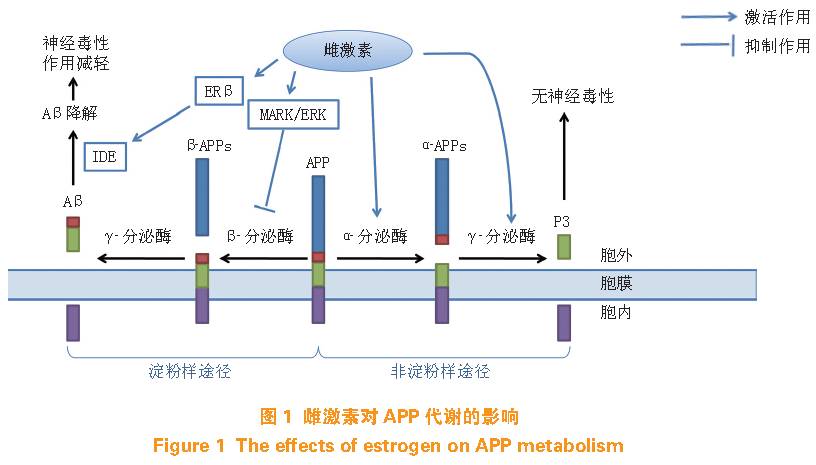
淀粉样前蛋白(
amyloidprecursor protein, APP
)的代谢可通过
2
种竞争性途径:淀粉样途径经
β-
分泌酶和
γ-
分泌酶生成
β-APPs
及
Aβ40/Aβ42
淀粉样片段;或非淀粉样途径经
α-
分泌酶生成具有神经保护作用的
α-APPs
及一些非淀粉样片段。雌激素对
APP
代谢的影响如图
1
所示,可通过激活
MARK/ERK
途径,减少
β-
分泌酶的表达,抑制
Aβ
的生成并促进
APP
的非淀粉样代谢途径,同时,通过刺激小胶质细胞的吞噬和降解作用,并调控
Aβ
的降解酶,促进
Aβ
的清除,其中,胰岛素降解酶(
insulin degrading enzyme, IDE
)是
Aβ
降解中的关键酶,
AD
动物脑内的
IDE
表达水平随年龄增长而逐渐减少,而雌激素可通过
ERβ
增加
IDE
的表达,促进
Aβ
的降解,减少
Aβ
沉积,保护神经元免受神经炎症及神经毒性损伤。与此同时,雌激素可改善由
Aβ
引起的神经毒性作用,淀粉样代谢途径生成的可溶性
Aβ
会诱发线粒体的裂变、减慢其活动并减少其氧化磷酸化,加重认知障碍,而雌激素可在激活
ERβ
后,与线粒体激酶锚定蛋白(
A kinase anchoringprotein 1,AKAP1
)结合,通过
PKA/CREB
通路促线粒体氧化磷酸化生成
ATP
,具有一定的线粒体保护作用。
突触可塑性是突触结构和功能的可变性,是学习记忆功能的主要神经生理学基础,雌激素可通过调节
CNS
突触可塑性改善绝经后
AD
患者的认知功能障碍。
Woolley
等
首先发现,处于动情周期动物的海马
CA1
区中内源性雌激素的水平可影响树突结构的复杂性。绝经后女性雌激素的缺乏会导致树突棘的减少,增高
AD
的患病风险。
17
β
E2
可通过对
NMDA
受体依赖机制的调控作用逆转树突棘的减少,并修复海马结构。研究常采用
OVX
动物模拟绝经后女性的雌激素缺乏状态,
OVX
野生型动物在接受
ERβ
激动剂治疗后,突触可塑性标志蛋白突触后致密蛋白(
postsynapticdensity protein 95, PSD-95
)和谷氨酸受体
-1
(
glutamatereceptor 1, GluR1
)的表达显著增加,有趣的是,在给予
ERα
激动剂的
OVX
野生型小鼠和
ER
β
基因敲除小鼠中未观察到此变化,提示雌激素对突触可塑性的影响可能具有受体选择性。
3
雌激素在帕金森病中的作用
PD
是由多巴胺能神经元的不可逆性或选择性退化引起的神经系统病变,表现为患者的运动功能异常迟缓。尽管目前常用的治疗药物较多,但现阶段治疗
PD
的药物仅限于对症治疗,并不能阻止
PD
病程的发展。研究发现,雌激素对帕金森病具有神经保护作用,并对多巴胺能神经元具有改善作用,而
ER
可能是抗帕金森病的一个潜在治疗靶点。
帕金森病病理特征之一是小胶质细胞介导的神经炎症,雌激素可通过结合
ER
,抑制脂多糖诱导分泌的炎症因子生成,发挥神经保护作用。同时,雌激素可通过抑制内向整流钾离子通道
Kir2.1
调控小胶质细胞活力,减少炎症发生,值得注意的是,雌激素的保护作用可被雌激素受体拮抗剂阻断,由此证明,
ER
是雌激素发挥抗神经炎症作用及神经保护作用中的关键因素。
ERα
可通过调控
PI3K
通路发挥抗炎效应,阻断
NF-κB
通路及其向核内的转移。在四氢吡啶(
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine,MPTP
) 诱导的
PD
动物模型实验中发现,雌激素还可抑制小胶质细胞活性并改善多巴胺能神经元缺失。
雌二醇对帕金森病多巴胺能神经元的保护作用与神经营养因子有关,可协同胶质细胞源性神经营养因子(
glial derived neurotrophic factor, GDNF
)经
N
-
钙黏蛋白的磷酸化
Y860
激活
PI3K/Akt
通路,保护多巴胺能神经元。体内实验发现,雌激素与
IGF-1
具有相互影响的作用,此过程通过
PI3K
的
P85
催化亚基调控完成,
IGF-1
可激活
PI3K/Akt
途径影响抗凋亡蛋白
Bcl-2
及凋亡蛋白
Bax
、
BAD
的表达,从而发挥促神经细胞生长分化及改善多巴胺能神经元功能的作用。
研究还发现,
17
β
E2
可激活
MAPK/ERK
通路活性,对促凋亡的
BAD
蛋白及可诱发纹状体神经元毒性的
GSK3β
蛋白表达具有抑制作用,且上述细胞信号通路基于
parkin
基因实现。同时,雌激素还可通过
PI3K/Akt
信号传导途径抑制
GSK3β
的生成,从而
GSK3β
对
tau
蛋白的磷酸化减少,神经纤维缠结及细胞凋亡的发生减弱。虽然
ERα
和
ERβ
在纹状体中均存在分布,然而
ER
α
基因敲除小鼠对
MPTP
诱导的纹状体多巴胺能损伤相比
ER
β
基因敲除小鼠更为敏感,推断
ERα
对帕金森病的调控作用更为重要。其他研究也相继发现,
ERα
特异性激动剂
PPT
对帕金森病的保护作用优于特异性
ERβ
激动剂
DPN
。
4
雌激素在多发性硬化症中的作用
MS
是由
Th1
、
Th17
细胞触发的
CNS
自身免疫性疾病,主要病理学表现为
CNS
多灶性脱髓鞘、炎症反应及轴突损伤。因实验性自身免疫性脑脊髓炎(
experimental autoimmune encephalomyelitis,EAE
) 动物在病理学表现上与人类
MS
具有诸多方面的相似性,所以
MS
研究常采用
EAE
动物模型作为实验对象。研究发现,雌激素对
EAE
具有以免疫调节为主、非抗炎效应为辅的保护作用。
Th1/Th2
的平衡对免疫系统疾病的发生发展具有重要作用,而
EAE
的病理过程以细胞免疫为主,主要由
Th1
细胞介导。雌激素经受体依赖途径,对
CD4
+
T
淋巴细胞的分化的调控作用具有浓度依赖性,低浓度雌激素可促
Th1
细胞应答;高浓度雌激素可减少
Th1
细胞分泌
IFN-γ
、
TNF-α
、
IL-12
等细胞因子,增强
Th2
细胞功能,使免疫反应向
Th2
转化,调控
Th1/Th2
平衡,减慢
MS
的发展进程。体外实验发现,雌激素可结合膜受体,经
PD-1/PD-L1
通路,减少促炎因子










