
第一作者:蒋雨恒,范英英,刘晓龙,谢君
通讯作者和单位:唐智勇,国家纳米科学中心
原文链接:https://doi.org/10.1021/jacs.4c03083
关键词:负载型催化剂,甲烷,光催化,甲酸
甲烷光氧化制备甲酸能够同时生成增值化学品并利用可再生能源。高产率和高选择性的甲酸生成是一个挑战,这主要是由于甲烷活化后的中间体转化和脱附的调控复杂性。在本研究中,我们使用第一性原理计算作为综合指导工具,发现通过精确控制贵金属助催化剂上的氧气活化过程以及金属氧化物载体上含碳中间体的吸附强度,可以精细调控甲烷光氧化产物的选择性。包含钯(Pd)纳米颗粒和单斜晶系氧化钨(WO
3
)的双功能催化剂(Pd/WO
3
)能够具备最佳的氧气活化动力学和中间体的氧化/脱附障碍,从而促进甲酸的形成。实验结果显示,该Pd/WO
3
催化剂在室温下使用分子氧并在全光谱光照射下,达到了4.67
mmol g
cat
−1
h
−1
的优异甲酸产率,并具有62%的高选择性。这些结果显著优于在相同条件下运行的最先进的光催化系统。
CH
4
在可持续发展中是双刃剑。一方面,由于储量丰富,甲烷是一种清洁的化石能源,另一方面,CH
4
是最常见的温室气体之一,在一个世纪内其全球变暖潜能是CO
2
的25倍。
以太阳能为动力的CH
4
光氧化技术为生产C1大宗化学品提供了一种很有前景的方法
,可以满足逐渐增长的能源利用和环境保护的需求。光催化CH
4
氧化领域的最新进展实现了CH
3
OH、HCHO和HCOOH等高附加值的液体化学品的生产。在这些产品中,
HCOOH具有巨大的市场价值
,是一种重要的工业产品,具有多种用途和不断增长的需求。
通过大量的试错实验已经实现了CH
3
OH和HCHO的选择性生成,其中通过改变BiVO
4
量子点的反应条件或在相同的反应条件下控制Au/In
2
O
3
催化剂中Au的大小,成功地实现了选择性调控。与此形成鲜明对比的是,
具有高产率和良好选择性的HCOOH生产的报道仍然很少
。例如,研究发现FeN
x
/C是一种具有竞争力的催化剂,可使用H
2
O
2
作为氧化剂生产HCOOH(1.165 mmol g
cat
-1
h
-1
)。然而,H
2
O
2
的高成本限制了其大规模实际应用。由于价格低廉且氧化能力温和,O
2
是一种理想的工业氧化剂。遗憾的是,用O
2
替代H
2
O
2
通常会导致HCOOH产率和选择性降低,
亟需开发在室温下使用O
2
直接高效光催化转化甲烷为甲酸的方法
。
在室温下和H
2
O溶液中,利用金属氧化物支撑的贵金属催化剂进行光催化CH
4
选择性有氧氧化被认为是一种极具吸引力的途径。一系列含氧化合物的形成是热力学上有利的:CH
4
®
CH
3
OH
®
HCHO
®
HCOOH
®
CO
2
(
示意图1
)。
要选择性地生成HCOOH,需要促进催化剂表面的CH
3
OH和HCHO继续氧化,同时防止HCOOH过度氧化为CO
2
。但这一严格要求很难满足因为
HCOOH的生成是一个多电子和多自由基反应,动力学缓慢,而形成的HCOOH在催化剂表面具有多个吸附基团和稳定的构型,因此容易被进一步氧化为CO
2
。
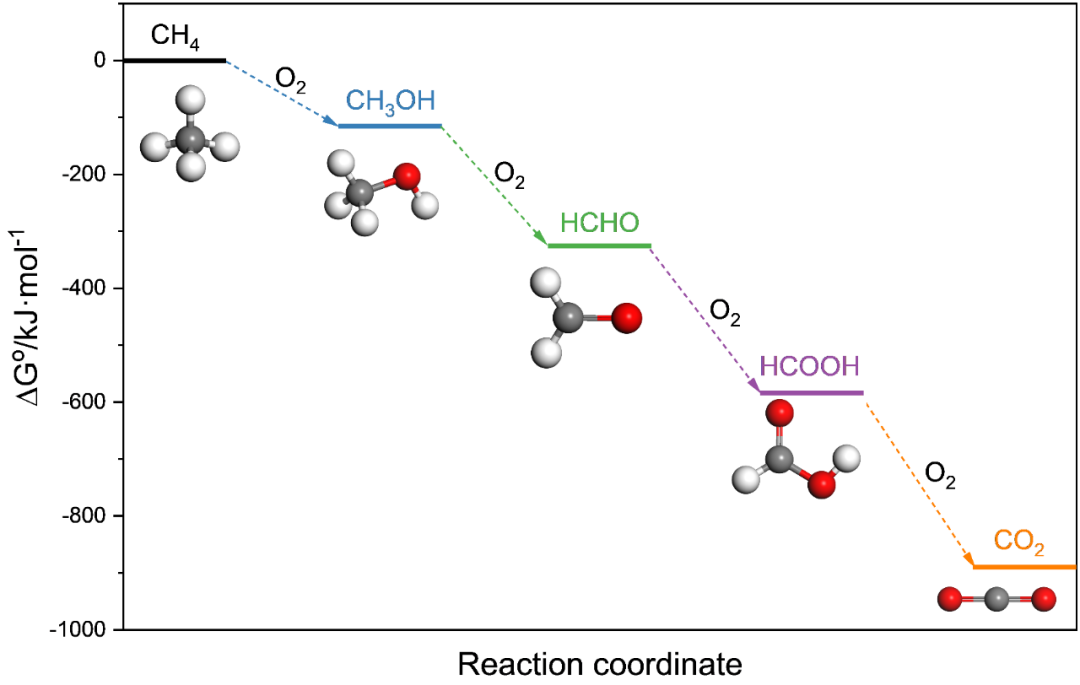
Scheme 1
. Standard molar Gibbs free energy corresponding to
continuous oxidation of methane to CO
2
at 298K.
在这项工作中,旨在通过利用贵金属和金属氧化物的协同效应来解决这一难题,从而同时优化中间体的转化和脱附过程,以选择性生成HCOOH。为了追求一种创新的方式,最大程度地减少试错带来的资源的使用和废物的产生,本工作采用了一种基于理论计算的方法来设计负载型催化剂。本工作的研究表明,贵金属具有适度的O
2
活化能力,有助于促进在金属氧化物表面的CH
4
及其中间体的转化,同时中间体适当的吸附和脱附能量有利于通过CH
4
氧化生成HCOOH。
理论预测高效光催化剂
为了寻找适合生产甲酸的光催化剂,我们收集了56篇关于使用负载贵金属的金属氧化物进行甲烷光催化氧化生成含氧化物的最新文献。从这些文献中可以看出,具有适当能带结构的TiO
2
、WO
3
和ZnO是最常用的金属氧化物,而Au、Pd和Pt则是由于其作为电子受体的匹配功函数而经常使用的贵金属。在这三种贵金属中,Au被认为具有较弱的O
2
活化能力,更倾向于形成甲醇(CH
3
OH)和甲醛(HCHO)而非甲酸(HCOOH)。为了探讨不同金属氧化物(TiO
2
、WO
3
和ZnO)和贵金属(Pt和Pd)组合对甲烷氧化选择性的影响,我们采用密度泛函理论(DFT)方法全面研究了反应路径及能量分布。
首先,我们研究了在贵金属表面上O
2
的活化及其后续活性氧物种(ROS)的形成(图1a)。在Pd基催化剂中,*O是含碳中间体夺取氢的主要氧化剂。当采用Pt (111) 时(图4.3d右栏),*OH成为含碳中间体夺取氢的主要氧化剂。作为CH
4
转化过程中的O
2
活化剂,Pt比Pd具有更强的氧化能力。本工作对各种金属氧化物(WO
3
、ZnO和TiO
2
)的表面进行了CH
4
氧化反应过程中的能量变化的计算(见图1b-f)。利用表面中间产物氧化和脱附的能量差作为描述符,来预测CH
4
氧化的选择性。
根据图1f中的数据,预测Pd/WO
3
是选择性生产HCOOH的最优催化剂(图1c)。产物的选择性受金属氧化物载体的影响。当WO
3
被ZnO取代时,HCHO成为最主要的产物(图1d)。在Pd/TiO
2
系统中(图1e),碳氢化合物中间产物与表面的结合力较弱,最终产物为CH
3
OH。贵金属催化剂在调节O
2
活化方面起着关键作用,而O
2
活化会影响产物选择性(图1f)。在WO
3
系统中,当*OH为主要氧化剂时,用活性更高的Pt替代Pd会促进*OCHO中间体的进一步氧化,并带来CO
2
生成量的增加(图1f)。不同的是,对于Pt/ZnO和Pt/TiO
2
系统,强烈的脱附趋势超过了在Pt表面产生的就有更强氧化性*OH物种的影响(图1f)。因此,与Pd作为助催化剂的体系相比,主要产物保持不变。
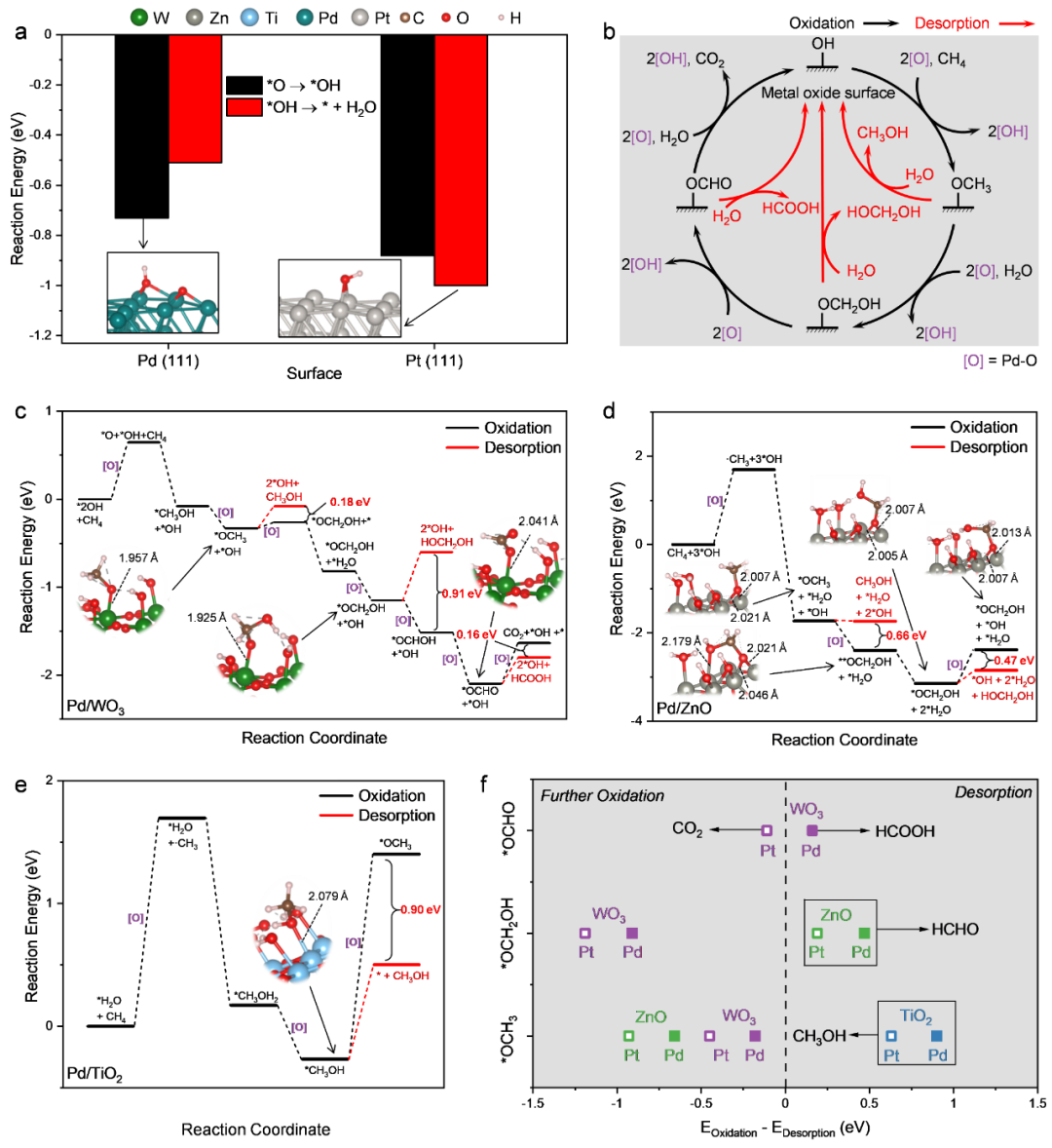
Figure 1.
Theoretical calculations of supported catalysts for
photocatalytic methane oxidation.
(
a
) Oxidation energy of ROS on
Pd (111) and Pt (111) surface, and configuration of dominant active
species involving methane oxidation is shown in insets.
(
b
)
Proposed
methane oxidation pathway, where [O] is the active species from O
2
activation on Pd. The detailed energy evolution of methane oxidation on
(
c
)
Pd/WO
3
, (
d
)
Pd/ZnO and (
e
) Pd/TiO
2
,
and insets present the corresponding intermediate configurations.
f
.
Energy diagram of competition between further oxidation and desorption.
催化剂的合成与表征
根据DFT计算结果,通过简单的硼氢化钠(NaBH
4
)冰辅助还原法,合成了克级的单斜相WO
3
支撑Pd的光催化剂,Pd的理论重量百分数为0.25
wt.%(Pd
0.25
/WO
3
)。如图2a显示,Pd
0.25
/WO
3
的X射线衍射峰与WO
3
的标准单斜相(PDF#43-1035)十分吻合。TEM图像(图2b)显示,平均直径为5.58 nm的球形Pd纳米颗粒随机分布在WO
3
表面。ACHAADF-STEM图像(图2c)显示的晶格间距分别为0.383 nm和0.225 nm,分别对应于WO
3
(002)晶面和Pd(111)晶面。EDS元素分布图证实了Pd(粉红色)纳米颗粒的存在以及O(蓝色)和W(橙色)元素的均匀分布(图2d)。
本工作还合成并表征了Au
0.25
/WO
3
,
Pt
0.25
/WO
3
、Pd
0.25
/ZnO、Pt
0.25
/ZnO、Pd
0.25
/TiO
2
和Pt
0.25
/TiO
2
的对比样品。XRD图谱和XPS光谱显示合成的光催化剂结构完全符合DFT计算模型。
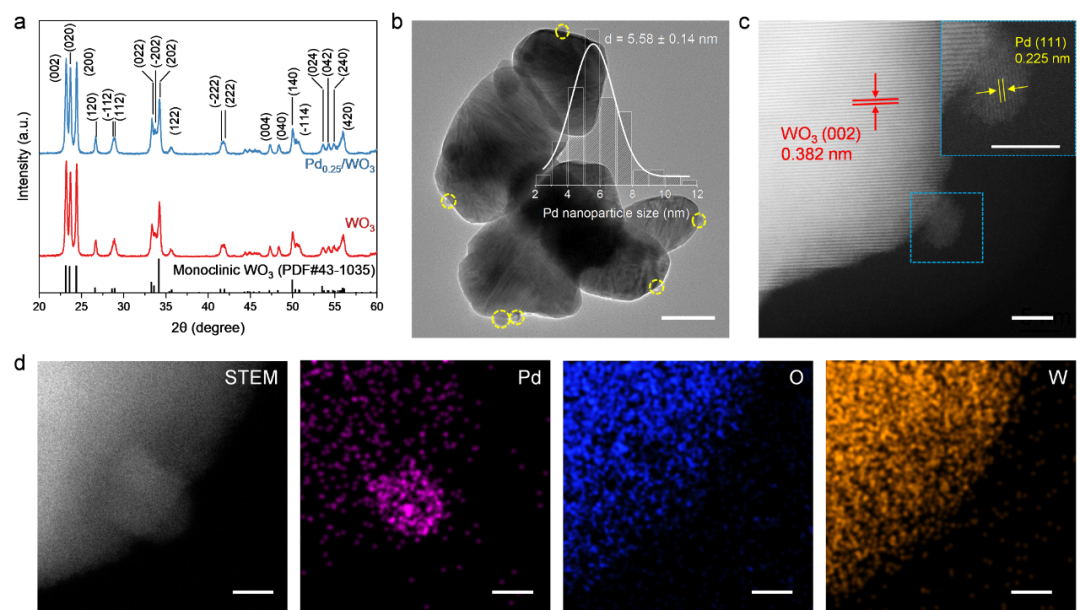
Figure 2.
Characterization of photocatalysts.
(
a
)
XRD patterns of Pd
0.25
/WO
3
and WO
3
.
(
b
)
Representative TEM image with scale bar of 50 nm, (
c
)
AC-HAADF-STEM and
(
d
)
EDS-mapping of
Pd
0.25
/WO
3
with scale bars of 5 nm. The yellow circles in
b denote Pd nanoparticles and the inset in b displays the size distribution of
Pd nanoparticles. The inset in c shows an enlarged view of the square area
marked in c with scale bars of 5 nm.
光催化甲烷氧化性能测试
在室温(25℃)和全光谱光照射下,在加压间歇反应器中评估了光催化CH
4
氧化的性能。比较了不同成分的贵金属负载金属氧化物的光催化CH
4
氧化性能。对于纯WO
3
,辐照5 h后只能检测到0.44 mmol g
cat
-1
的HCHO和CO
2
的混合物;而在相同的反应条件下,Au
0.25
/WO
3
的C1产物产量大幅增加,达到5.21 mmol g
cat
-1
。本工作注意到,Au
0.25
/WO
3
上的产物中CH
3
OH、HCHO和HCOOH的比例分别为12%、50% 和29%,这与Au具有较弱的O
2
活化能力的事实一致,更倾向于形成CH
3
OH和HCHO。因此,本工作特别关注作为助催化剂的Pt或Pd。不出所料,引入Pd或Pt助催化剂可以显著促进液态产物的形成(图3a)。5 h后,Pd
0.25
/WO
3
的HCOOH产量达到23.32 mmol g
cat
-1
,选择性为62%。而Pt
0.25
/WO
3
的CO
2
产量增加到3.67 mmol g
cat
-1
,HCOOH选择性降低到49%(图3a)。本工作还研究了金属氧化物载体的影响。在光照射下,Pd
0.25
/ZnO、Pt
0.25
/ZnO、Pd
0.25
/TiO
2
和Pt
0.25
/TiO
2
上的产物包括CH
3
OOH、CH
3
OH和HCHO(表图3a),但没有检测到HCOOH。Pd
0.25
/ZnO和Pt
0.25
/ZnO的主要产物是HCHO,而Pd
0.25
/TiO
2
和Pt
0.25
/TiO
2
的主要产物是CH
3
OH。所有这些催化结果都表明,Pd/WO
3
是产生HCOOH的最佳光催化剂,光催化CH
4
氧化的选择性由金属氧化物载体和贵金属助催化剂共同决定,这与DFT计算结果完全一致。
随着在WO
3
上Pd负载量的增加,光催化活性得到提高(图3b),在0.25 wt.%时含氧化合物产量最高。进一步增加Pd的负载量会导致含氧化合物产量略有下降,这可能是由于Pd纳米粒子的尺寸从5.58 nm增加到7.41nm和由此产生的屏蔽效应所致的。在没有O
2
的情况下,含氧化合物的产量从37.48 mmol g
cat
-1
急剧下降到0.31 mmol g
cat
-1
。更重要的是,在厌氧条件下检测不到CH
3
OOH和HCOOH,这表明光催化CH
4
氧化成CH
3
OOH和HCOOH需要O
2
。如图3c所示,含氧化合物的总生成量随反应时间的延长而逐渐增加,表明Pd
0.25
/WO
3
具有持续催化CH
4
氧化的能力。至于HCOOH的选择性,在最初的5 h内有所提高,达到最大值62%。当反应时间超过5 h后,过氧化产物CO
2
迅速增加,导致HCOOH的选择性略有下降。
简而言之,Pd
0.25
/WO
3
在5 h内的HCOOH生成速率和选择性分别高达4.67 mmol g
cat
-1
h
-1
和62%。这些数值明显高于室温下以O
2
为氧化剂的所有最先进光催化CH
4
氧化系统的性能(图3d),甚至超过了高温下的性能(橙色部分)。图3e列出了Pd
0.25
/WO
3
在不同单色光下的QE。QE的变化趋势与UV-vis DRS一致,揭示了反应是一个光催化过程。365 nm波长下的QE值最高,为5.39%,而更令人兴奋的是,420 nm波长下的QE值为2.98%,这表明Pd
0.25
/WO
3
催化的CH
4
氧化过程中具有可见光催化活性。
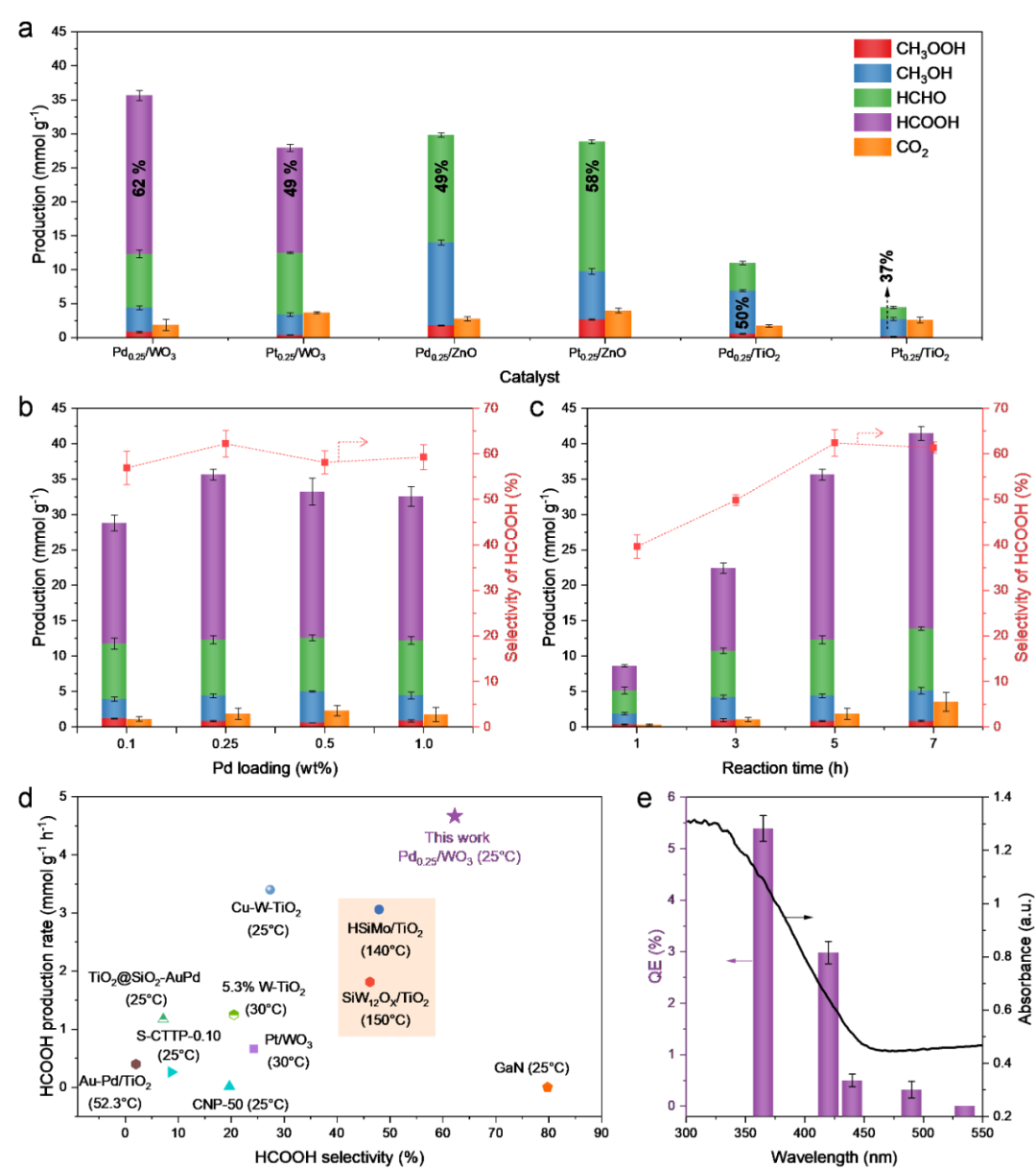
Figure 3.
Photocatalytic methane oxidation performance.
(
a
)
Photocatalytic
performance over different catalysts. Photocatalytic performance over (
b
)
Pd
x
/WO
3
with varied Pd amount, (
c
) Pd
0.25
/WO
3
for different reaction time.
(
d
)
Comparison of catalytic
activity for photooxidation of methane to HCOOH over Pd
0.25
/WO
3
with reported photocatalysts.
(e)
QE
values at varied monochromatic wavelengths along with UV-vis DRS of Pd
0.25
/WO
3
.
机理探究
除了光催化性能之外,通过实验表征对机理进行研究也将有力地验证DFT计算结果。为了深入了解光生载流子的分离和迁移,进行了超快瞬态吸收(TA)测量。在波长为350 nm的泵浦脉冲激发下,WO
3
和Pd
0.25
/WO
3
均展示出在450-750
nm范围内的负峰TA光谱(图4a-b)。这些负峰归因于基态漂白(GSB),反映了激发态的弛豫过程。Pd
0.25
/WO
3
中的载流子寿命明显长于WO
3
中的载流子寿命(图4c),表明Pd
0.25
/WO
3
的载流子分离效率优于WO
3
。此外,瞬态光电流测量显示Pd
0.25
/WO
3
的光电流强度高于WO
3
,表明Pd的存在增强了光生载流子的分离效率。原位XPS测试表明在光催化甲烷转化过程中,Pd纳米颗粒作为电子受体发挥作用。
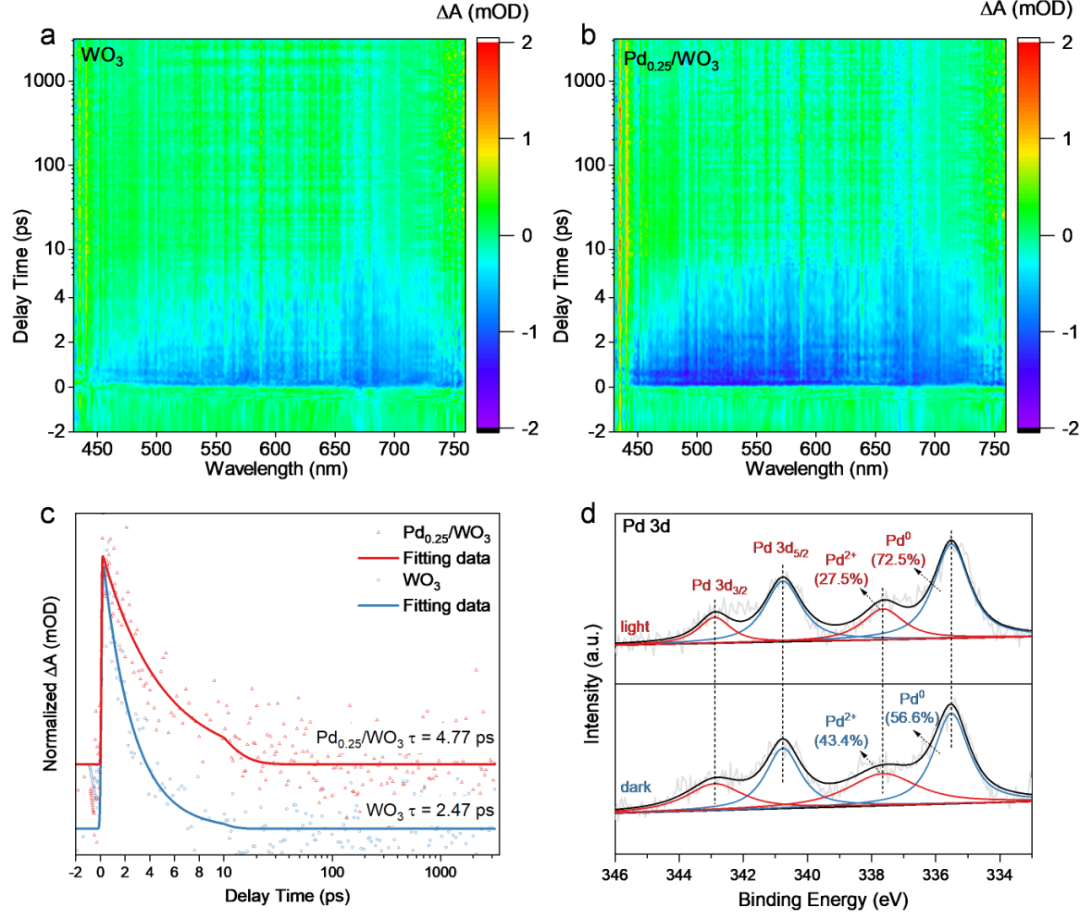
Figure 4. Carrier separation property.
2D mapping TA spectra of (
a
) WO
3
and (
b
) Pd
0.25
/WO
3
under 350 nm light irradiation.
(
c
)
Normalized decay kinetic curves in TA spectra of WO
3
and Pd
0.25
/WO
3
at 600 nm.
(
d
)
In situ
high-resolution Pd 3d XPS spectra of Pd
0.25
/WO
3
under
dark and irradiation condition.
分子O
2
活化是光催化CH
4
氧化过程的关键步骤。为理解贵金属表面的O
2
活化机制,本工作进行了原位DRIFTS分析,其对活性物种具有良好的灵敏度。当存在O
2
和H
2
O的光照条件下,Pd
0.25
/WO
3
的原位DRIFTS光谱在3430 cm
-1
和990 cm
-1
处显示出两个明显的峰(图5a)。根据文献,这两个峰分别对应于*OH和单氧(*O)中间物种。此外,*O的吸收峰强度超过了*OH的吸收峰强度(图5a),这表明*O是Pd
0.25
/WO
3
上驱动CH
4
活化和转化的主要氧化剂。相反,在相同条件下,Pt
0.25
/WO
3
的原位DRIFTS光谱显示出一个突出的*OH吸收峰和一个非常弱的*O峰(图5b),这表明*OH在Pt
0.25
/WO
3
上是主要的氧化剂。在Ar和H
2
O的气氛下加入光照,只观察到归属于H
2
O氧化生成的*OH信号(图5),Pd
0.25
/WO
3
和Pt
0.25
/WO
3
上的*OH峰强度分别是O
2
存在条件下的~50%和~20%。所有这些DRIFTS测试结果表明,WO
3
上的Pd纳米粒子和Pt纳米粒子是O
2
的活化位点,它们分别通过O
2
还原反应生成*O和*OH的活性物种。自由基淬灭实验和原位ESR提供了进一步的证据,表明由O
2
还原生成的ROS主要作为氧化剂驱动甲烷活化和转化,这些发现与理论计算结果一致。
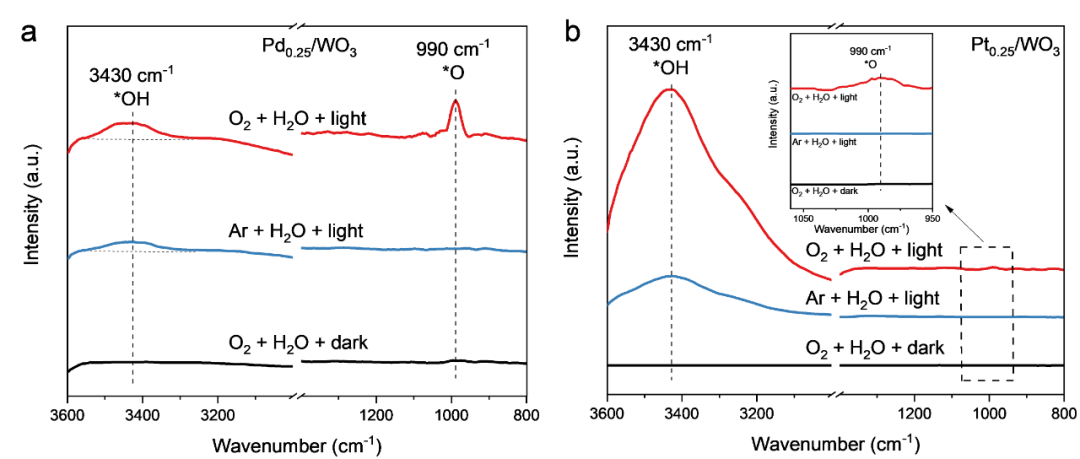
Figure 5. Intermediates characterization of O
2
activation.
In situ
DRIFT spectra of (a) Pd
0.25
/WO
3
and
(b) Pt
0.25
/WO
3
in the presence of O
2
and H
2
O
or Ar and H
2
O in darkness and under light irradiation.
原位DRIFTS被用于进一步探究光催化CH
4
氧化的反应路径,在CH
4
、O
2
和H
2
O存在的光照情况下收集信号(图6)。在Pd
0.25
/WO
3
表面,可以在3000-2800
cm
-1
范围内明显地发现吸附的CH
3
OH的*CH
3
信号和吸附的HCHO的*CH
2
信号,以及吸附的HCOOH的C=O伸展振动在1720 cm
-1
处的强信号(图6a),这表明在Pd
0.25
/WO
3
表面有利于CH
4
通过CH
3
OH和HCHO中间体氧化为HCOOH。值得注意的是,虽然形成了过氧化中间产物*OCO,但没有检测到CO
2
气体信号(图6b)。相反,当Pt取代WO
3
表面上的Pd时,可以观察到明显的CO
2
信号。此外,与Pd
0.25
/WO
3
相比,Pt
0.25
/WO
3
的*HCOOH与*OCO的强度比要低得多,这表明Pt纳米粒子的引入会导致*HCOOH过度氧化为CO
2
,而WO
3
表面的Pd纳米粒子则有助于选择性地形成HCOOH。
当WO
3
被其他金属氧化物取代时,Pd
0.25
/TiO
2
和Pd
0.25
ZnO表面没有吸附的HCOOH的信号(图6c-f)。对于Pd
0.25
/TiO
2
(图6c和d),在1900-1200 cm
-1
区域,吸附的CH
3
OH(*OCH
3
)峰比吸附的HCHO(*CH
2
)峰强度更高,表明表面吸附的CH
3
OH不会被进一步氧化,有利于CH
3
OH的生成。以ZnO为载体时(图6e和f),在3000-2800 cm
-1
范围内,*CH
2
信号比*CH
3
信号强得多,证明ZnO表面通过氧化吸附的CH
3
OH促进了HCHO的生成,并抑制了HCHO的过度转化。总之,原位DRIFTS表征结果表明了金属氧化物以及负载的贵金属的合理组合可有效调节中间产物的吸附和转化,从而控制产物的选择性。
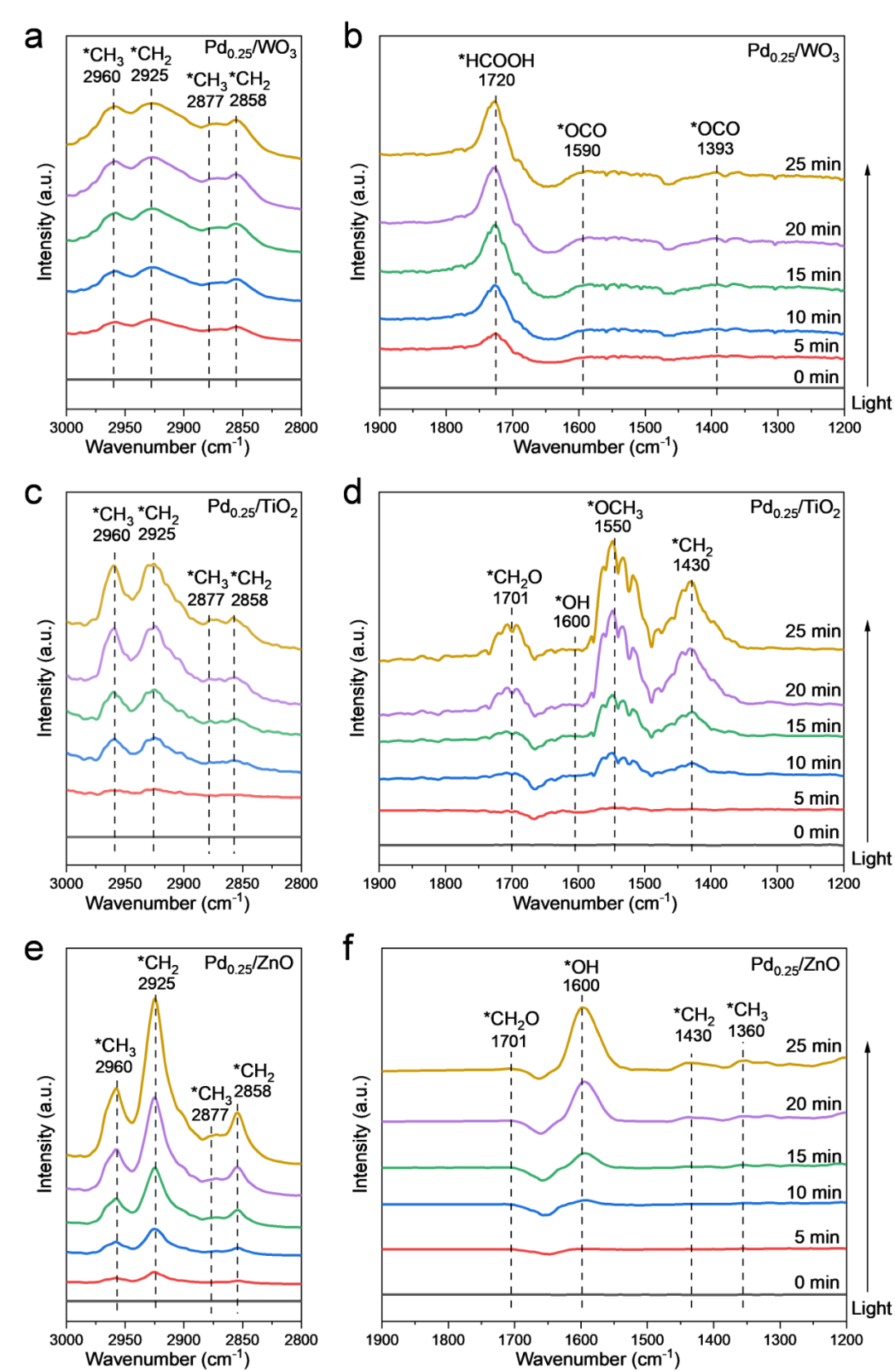
Figure 6. Intermediates characterization of photocatalytic
methane oxidation.
In situ
DRIFT spectra of
(
a
,
b
)Pd
0.25
/WO
3
, (
c
,
d
) Pd
0.25
/TiO
2
and (
e
,
f
) Pd
0.25
/ZnO in the presence of CH
4
,
O
2
and H
2
O for different light irradiation time.
本工作建立了一种合理设计光催化剂的范例,能够在温和条件下高效选择性地氧化甲烷。基于理论计算,我们预测并实现了Pd/WO
3
光催化剂,通过精确评估中间体进一步氧化和解吸之间的能量差异来最大化甲酸(HCOOH)的产率,这些能量差异分别由活性氧物种和吸附构型决定。我们相信这一策略将大大减少经验性的试错需求,加快高性能催化剂在包括但不限于甲烷转化等挑战性反应中的探索。
[1]
J Am Chem Soc
2024,
146
(23), 16039.
[2]
Nature Sustainability
2021, 4 (6), 509.
[3]
Angew. Chem. Int. Ed.
2024, e202404658.
[4]
J Am Chem Soc
2022, 144 (35), 15977.
[5]
J Am Chem Soc
2023, 145 (4), 2698.
[6]
Nat. Commun.
2024,
15
, 4679.
[7]
CCS Chemistry
2023, 5 (1), 30.









