下面是PCR设计的一些基本原则以及引物设计的方法步骤与资料,希望对于搞实验的同鞋有所帮助。
一、PCR引物设计原则
1、引物长度一般在15-30bp。
引物长度一般为15-30bp,常用的为18-27bp,但不应大于38bp,因为过长会导致其延伸温度大于74℃,不适于Taq DNA聚合酶进行反应;
2、引物GC含量一般为40%-60%。
引物GC含量一般为40%-60%,以45-55%为宜,GC含量过高或过低都不利于引发反应。上下游引物GC含量和Tm值要保持接近;
3、引物所对应的模板序列的Tm值最好在72℃左右。
Tm值在72℃左右可使复性条件最佳,至少要在55-80℃之间。Tm值的计算有多种方法,如按公式Tm=4(G+C)+2(A+T),在Oligo软件中使用的是最邻近法(the nearest neighbor method);
4、引物3’端的碱基一般不用A。
引物3’端的碱基一般不用A,因为A在错误引发位点的引发效率相对比较高。
5、引物3’端出现3个以上的连续碱基,如GGG或CCC,也会使错误引发机率增加。
6、引物3’端的互补、二聚体或发夹结构也可能导致PCR反应失败。
7、如扩增编码区域,引物3’端不要终止于密码子的第3位,因密码子的第三位易发生简并,会影响扩增特异性与效率;
8、引物5’端可以修饰。
引物5’端序列对PCR影响不太大,因此常用来引进修饰位点或标记物。
9、碱基要随机分布,且引物自身和引物之间不能有连续4个碱基的互补。
引物序列在模板内应当没有相似性较高,尤其是3’端相似性较高的序列,否则容易导致错误引发。降低引物与模板相似性的一种方法是,引物中四种碱基的分布最好是随机的,不要有聚嘌呤或聚嘧啶的存在。引物自身不应存在互补序列,否则引物自身会折叠成发夹结构(Hairpin)使引物本身复性。这种二级结构会因空间位阻而影响引物与模板的复性结合。引物自身不能有连续4个碱基的互补。两引物之间也不应具有互补性,尤其应避免3’端的互补重叠以防止引物二聚体(Dimer与Cross Dimer)的形成。引物之间不能有连续4个碱基的互补。引物二聚体及发夹结构如果不可避免的话,应尽量使其△G值不要过高(应小于4.5kcal/mol)。否则易导致引物二聚体带的 产生,并且降低引物有效浓度而使PCR反应不能正常进行。
10、引物的ΔG值最好呈正弦曲线形状,即3’端ΔG值较低,而5’端和中间ΔG值相对较高。
ΔG值(自由能)反应了引物与模板结合的强弱程度,一般情况下,引物的ΔG值最好呈正弦曲线形状,即3’端ΔG值较低,而5’端和中间ΔG值相对较高。3'端的ΔG值相对要低,且绝对值不要超过9,引物的3’端的ΔG值过高,容易在错配位点形成双链结构并引发DNA聚合反应。
11、引物应在核酸保守区内设计并具有特异性。引物与非特异扩增序列的同源性不要超过70%或有连续8个互补碱基同源。
二、Real Time PCR引物设计原则
Real Time PCR用引物与普通PCR引物设计要求不同。RealTime PCR是让目的基因按照理论值进行扩增,对非特异性反应和引物二聚体要求严格。
Real Time PCR引物设计原则
|
扩增片段大小
|
★
|
80-150 bp(尽量限制在300 bp以内)
|
|
|
Primer长度
|
★
|
17-25 base
|
|
|
GC含量
|
★
|
40-60%(最好45-55%)
|
|
|
Tm值
|
★ ★ ★
|
两条引物的Tm值尽量接近,应用专用软件计算Tm值
|
|
|
序列
|
★
|
整体上碱基不能过偏 个别部分避免GC rich或AT rich(特别是3’端) 避免T/C连续,A/G连续
|
|
|
|
|
3'末端序列
|
★
|
3’末端避免GC rich或AT rich
3’末端碱基最好为G 或C
3’末端碱基尽量避免为T
|
|
|
互补性
|
★ ★ ★
|
引物内部或两条引物之间避免3 base以上的互补序列
引物3’末端避免2 base以上的互补序列
|
|
|
特异性
|
★ ★ ★
|
使用BLAST检索,确认引物特异性
|
|
|
RT-PCR用引物
|
★ ★ ★
|
尽量在Exon junction上设计引物,限制基因组DNA扩增
|
|
三、常用的引物设计软件
“Primer premier”,“Oligo”,“Vector NTI Suit”,“DNAsis”,“Omiga”和“DNAstar”。
下面详细介绍一下Primer premier 5.0的用法
1、通过File下的New或Open载入需要设计引物的序列

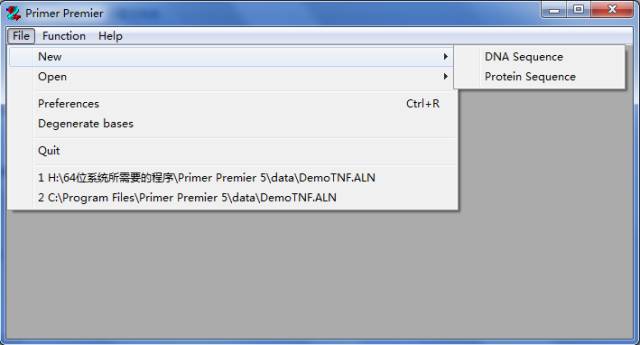
2、进入到程序的引物设计窗口
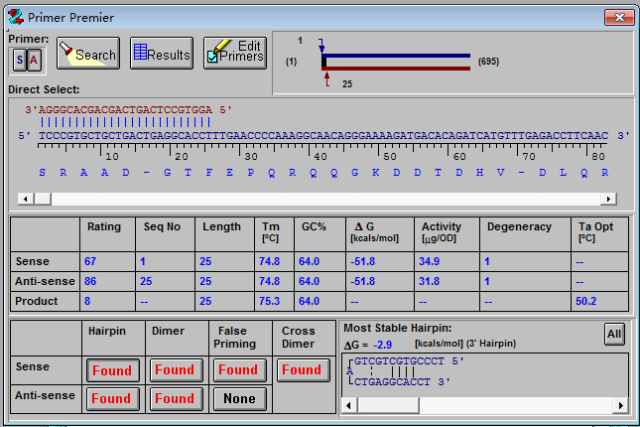
3、点击search,则出现新的界面
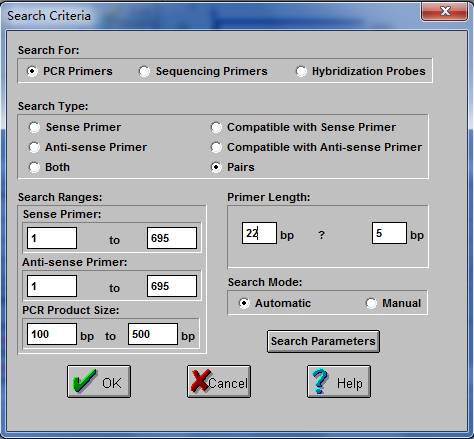
在新界面设置你需要设计的引物的相关参数。主要有五个要设置的地方。第一个地方是选择设计PCR引物,测序引物和杂交探针,如果要做PCR就选第一个圈圈“PCR Primers”。第二个地方是搜寻模式,一般我们搜索引物是以对搜索,所以一般选"pairs"。第三个地方是正负链的所在区间以及产物的大小长度,这个按自己的需要来,跟实验目的有关,具体情况具体分析。第四个地方是引物的长度以及正负波动值,一般来说引物长度在18-27范围内。第五个地方是选择模式,一般选“Automatic”。设置完上面所说的这些地方后按“OK”键,则跳转到新界面。
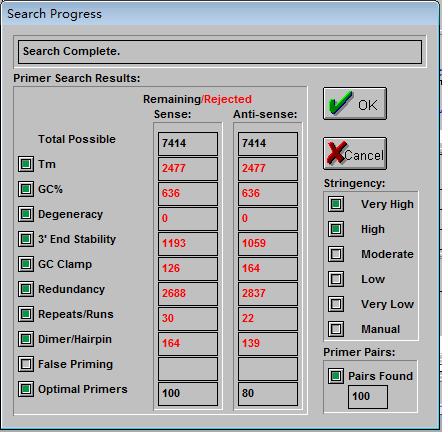
4、点击“OK”,出现如下新界面
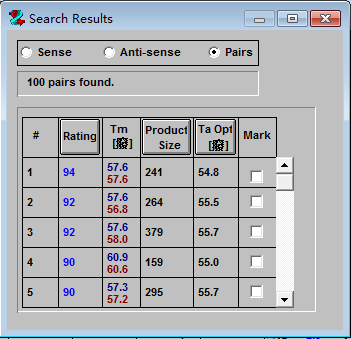
显示结果按照评估分数从高到低向下排列, 一般选取评估分数较高的结果。打分是衡量引物质量的综合性参数,利用打分系统可以对引物进行有效评估和引物间进行对比选择提供有效的证据。如果我们只想要寻找正向或反向的引物,就可以选择“Sense”或“Anti-sense”按钮。
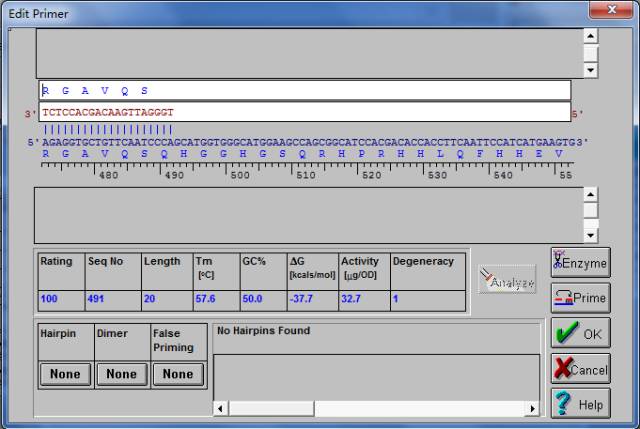
5、点击评分高的结果,就可以显示引物的详细信息,如下图。
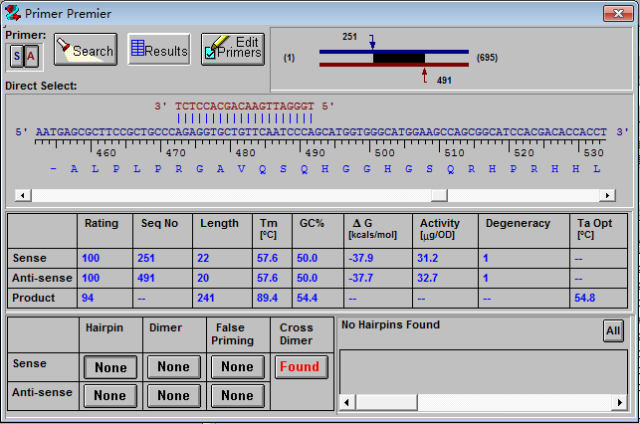
该图左上角的两个按钮
 和
和
 分别代表的是正向和反向引物。该图中间部分给出了引物的得分,位置,长度,Tm值,GC含量,自由能等信息。该图最下面的模块给出了正反引物是否形成发夹结构,二聚体,错配以及引物之间的二聚体等情况,红色代表有。点击
分别代表的是正向和反向引物。该图中间部分给出了引物的得分,位置,长度,Tm值,GC含量,自由能等信息。该图最下面的模块给出了正反引物是否形成发夹结构,二聚体,错配以及引物之间的二聚体等情况,红色代表有。点击
 或
或
 我们会看到出现这些结构的具体信息,比如开始的位置,产生的自由能。6、如果我们对搜索到的引物不满意,可以手动调整引物的位置,同时可以点击
我们会看到出现这些结构的具体信息,比如开始的位置,产生的自由能。6、如果我们对搜索到的引物不满意,可以手动调整引物的位置,同时可以点击
 的按钮对引物进行修改。如下图,我们可以增加,删除或修改碱基。直到找到最佳的结果。
的按钮对引物进行修改。如下图,我们可以增加,删除或修改碱基。直到找到最佳的结果。