前几期的博文中我们已经介绍了质量检查和质量控制的基本内容,本期我们来看一下重测序/外显子测序分析的经典流程——BWA MEM + GATK,本期的内容包含分析代码,分析过程中可能会遇到的问题以及我个人的一些经验。如果大家喜欢,欢迎分享到朋友圈。
RAW reads QC
Trimmomatic=/your/path/to/trimmomatic-0.36.jar
adapter=/your/path/to/adapters
java -jar "$Trimmomatic" PE -phred33 ${name}_1.fq.gz ${name}_2.trim.fq.gz ${name}_R1.clean.fq.gz ${name}_R1.unpaired.fq.gz ${name}_R2.clean.fq.gz ${name}_R2.unpaired.fq.gz ILLUMINACLIP:"$adapter"/Exome.fa:2:30:10:1:TRUE LEADING:20 TRAILING:20 SLIDINGWINDOW:4:15 -threads 2 MINLEN:36
关于本期博文所用软件的安装方法请见文末链接;
上一期的博文已经包含了对 Trimmomatic 的介绍,这里就不在赘述了。
Prepare reference genome
samtools faidx ref.fa
bwa index -a bwtsw ref.fa
Picard=/your/path/to/picard.jar
java -jar $Picard CreateSequenceDictionary REFERENCE=ena.fasta OUTPUT=ref.dict
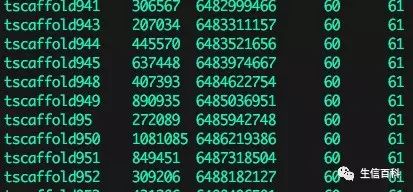
运行 samtools faidx ref.fasta 将得到一个 ref.fa.fai 的文件,这个文件会被 BWA 和 GATK 调用,如果文件不存在软件会报错。文件中的每一行为一个 contig 的记录,包含:contig name, size, location, basesPerLine, bytesPerLine,如下图所示:
运行 bwa index -a bwtsw ref.fa 将得到 ref.fa.amb,ref.fa.ann,ref.fa.bwt,ref.fa.pac,ref.fa.sa 5个文件,这些文件是 BWA mapping 时所必须的。
对于比较大的基因组,比如火炬松基因组有 22G ,运行时间会超过 10 个小时 (同时也取决于 CPU 的计算能力)。
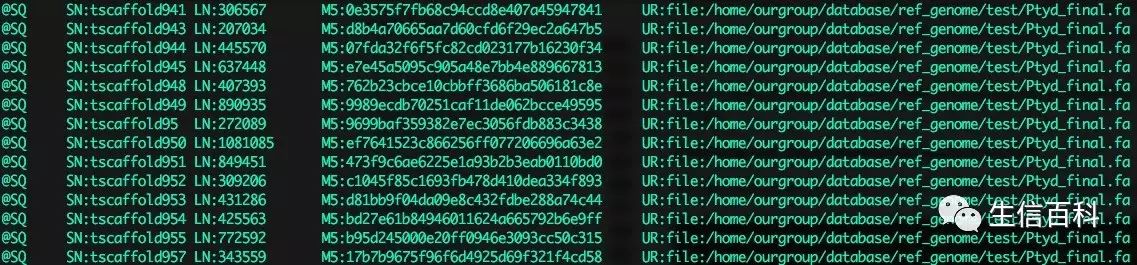
运行Picard 的 CreateSequenceDictionary 将得到 ref.dict,其描述参考基因组的内容,如下图所示:
对于比较大或者比较碎的基因组,比如火炬松 22G 基因组,会出现 java 运行错误,提示 java 虚拟机内存不足。这时,即使改变 java 运行参数 (比如设置 Xmx 等) 也不能正常运行,千万不要耗费时间在这里!
由于 .dict 文件只是 GATK 必须的,如果你的基因组很大不能建立 .dict, 可以考虑换成 BWA + Samtools + Bcftoos 流程,或者自己写代码生成 .dict 文件。
Mapping
ref=/your/path/to/ref.faecho "Job started "`date`" "bwa mem -M ${ref} ${name}_1.fq.gz ${name}_2.fq.gz -R "@RG\tID:${name}\tPL:Illumina\tSM:${name}\tLB:WGS" -t 12 | samtools view -Shb ${ref}.fai - > ${name}.PE.bam
bwa mem -aM ${ref} ${name}_R1.unpaired.fq.gz -R "@RG\tID:${name}\tPL:Illumina\tSM:${name}\tLB:WGS" -t 12 | samtools view -Shb "$ref".fai - > ${name}.R1.bam
bwa mem -aM ${ref} ${name}_R1.unpaired.fq.gz -R "@RG\tID:${name}\tPL:Illumina\tSM:${name}\tLB:WGS" -t 12 | samtools view -Shb "$ref".fai - > ${name}.R2.bam
samtools merge ${name}.merge.bam ${name}.PE.bam ${name}.R1.bam ${name}.R2.bamecho "Job ended "`date`" "
这里需要注意的有几点:
我们可以直接用 samtools 把 mapping 结果转成 bam 格式,不需要额外的运行步骤!
在 mapping 的时候可以直接用 -R 参数加 header,后面的分析就能正常进行了,可省略用 Picard 加 header 的步骤!
运行 BWA 的时候一定要申请足够的内存,并检查生成 bam 文件的大小。我遇到过 "运行内存不足不报错直接生成一个很小的 bam (几十M)" 的情况,正常的大小应该为几百兆!
我没有给运行时间的测试,因为大家研究的物种不同,参考基因组大小、测序深度也不一样,运行时间会有很大差异;而且不同的集群计算能力差异非常大,不同的集群运行速度可能会相差几倍!
Sort,Fix Mate Information and mark duplicates
Picard=/your/path/to/picard.jar
java -jar $Picard SortSam INPUT=${name}.merge.bam OUTPUT=${name}.sorted.bam SORT_ORDER=coordinate VALIDATION_STRINGENCY=SILENT
java -jar $Picard FixMateInformation INPUT=${name}.sorted.bam SO=coordinate
java -jar $Picard MarkDuplicates INPUT=${name}.sorted.bam OUTPUT=${name}.dedup.bam METRICS_FILE=metrics.txt VALIDATION_STRINGENCY=SILENT
java -jar $Picard BuildBamIndex INPUT="$sample".dedup.bam VALIDATION_STRINGENCY=SILENT
rm ${name}.PE.bam ${name}.R1.bam ${name}.R2.bam ${name}.merge.bam ${name}.sorted.bam
这里需要注意的是:
如果 Picard 总抱怨文件格式不对而不能运行,可以尝试加上 VALIDATION_STRINGENCY=SILENT 参数。
GATK 可以处理 Picard 标记为重复的 reads,因此可以仅做标记不删除。