

随着药监飞检的常态化,不断有药企被撤销或收回GMP证书,甚至被注销《药品生产许可证》,今年上半年就有25家制药企业消失了。在这25家企业里,存在的问题不一定都一样,但可以肯定的是,药企质量体系与GMP规范相差甚远。笔者选择其中的一家进行研究分析,供大家思考与借鉴。
重庆格瑞林药业有限公司于今年2月被重庆药监局注销了许可证。导火索是群众举报,2016年4月国家总局对格瑞林进行飞行检查,发现存在严重的违法违规行为,并收回该企业相关《药品GMP证书》。
冰冻三尺并非一日之寒,抽检发现该企业产品的不合格情况复杂,如果早点对症下药并及时整改,或许就不会发生被吊证的情况。
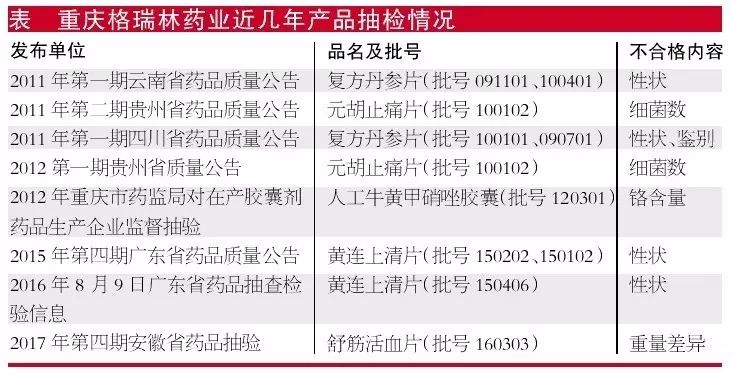
此次总局对格瑞林飞行检查发现的问题共8条,每条都很具有代表性,那些未被飞检或侥幸未被发现严重不合规的药企,真的需要好好自查了。本次检查发现的问题可以归纳为5种主要类型:
出租出借人工牛黄原料药生产资质。
按《药品管理法》第八十一条处罚。为了3%的收益,却要负100%的责任,相信不会有企业想这样干了。
涉嫌外购人工牛黄贴牌进行销售。
按《药品管理法》第七十九条处罚,违反2010年版GMP条款第4条。只要检查组查税、查账,估计很多人立刻会提心吊胆了。特别是中药饮片生产企业,因为品种多、检验成本高,加上生产技术含量不高,市场竞争激烈,还有产地的农副产品加工饮片、无证生产饮片等情况,导致企业合规成本高、有心无力。即便如此,触到红线,也就只有等着吊证了。
未严格按照《人工牛黄工艺规程》、人工牛黄药品注册申报资料规定的生产工艺生产人工牛黄。《人工牛黄工艺规程》未明确所用原辅料的来源、前处理方法和保障原辅料质量的措施;未按照人工牛黄工艺规程制定岗位操作规程。
违反《药品管理法》第十条,违反GMP条款第168条、150条等。该缺陷涉及注册工艺变更的问题,需要按照《药品注册管理办法》进行补充申请。
但是,申请变更工艺是否能获得药监局的批准,需要增加哪些试验和研究,需要多长时间能获批等问题,一直困扰着企业的决定。比如二十年前申请药品注册的工艺,由于当时的要求较低和条件所限,与现在的技术发展已不相适应,而要申请工艺变更难度极大,即使按照《关于开展药品生产工艺核对工作的公告(征求意见稿)》能给企业更正的机会,但代价也挺大。
期待好政策的同时,企业还是需要未雨绸缪,排查风险,首先做到目前在产品种必须严格按照注册工艺执行。
未对部分主要物料供应商进行审计。
违反2010年版GMP第255条。供应商资料不齐、证件过期,主要原辅料供应商未开展现场审计等,均是企业常见问题。
无论是否因供应商提供的原辅料、药包材质量的问题,如果产品不合格,最终负责的仍是制剂企业,何况现在药包材、药用辅料均实施关联审评,就是要求制剂企业对上游企业的质量情况进行全面控制。所以,任何流于形式的供应商审计都行不通,在供应商现场审计时,如果发现缺陷就应该要求企业改进,将风险控制到最小。
不能追溯人工牛黄原料药生产使用的物料的验收、入库、出库,以及物料供应商、生产商、批号、数量等信息。不能追溯人工牛黄原料药的入库、出库,以及批号、数量、销往单位等信息。不能从销售部门追查每批原料药人工牛黄销售情况。根据批生产记录不能追溯物料来源和流向。
违反了2010年版GMP条款第10条、第103条、第106条、第159条、第175条、第295条等。可见与物料相关的记录均不符合,整个追溯系统是崩溃的。物料管理看似简单,却是最容易出问题的环节,一旦第一步错就会步步错。对于检查人员来说,检查物料的方法太多了,可以现场抽查、库存台账抽查、批记录抽查、进货凭证抽查,正查、反查、对照查,上游供货查、下游客户查等,让任何不合规的地方无处遁形。
随着社会的进步、中国加入ICH,合规成为每个制药企业必须遵守的原则。外部飞检和内部举报投诉的双重压力下,企业必须做好内功,真正落实合规生产经营,方能降低合规风险。如此,企业才能不断提高和壮大,中国的制药行业才能有实力更多参与国际的竞争中。
(作者单位:CIO合规保证组织)
■编辑 余如瑾
★更多深度报道见《医药经济报》~ 









