来源:国家食品药品监督管理总局
2017年08月21日,国家食品药品监督管理总局发布抗菌药物药代动力学/药效学研究技术指导原则的通告(2017年第127号),
为进一步规范和指导我国抗菌药物研究,国家食品药品监督管理总局组织制定了《抗菌药物药代动力学/药效学研究技术指导原则》(见附件),现予发布。
特此通告。
附件:抗菌药物药代动力学/药效学研究技术指导原则
食品药品监管总局
2017年8月4日
抗菌药物药代动力学/药效学研究
技术指导原则
一、概述
(一)抗菌药物作用特点及临床试验要求
抗菌药物作用特点是杀灭或抑制入侵到机体内的外来病原菌而发挥药理效应,其疗效取决于抗菌药物、病原菌和机体三者相互作用的结果。病原菌种类复杂,致病力不同并可能存在不同的耐药机制,是致病关键因素;机体自身免疫功能可防御病原菌入侵,其功能正常或缺陷与否可影响抗菌治疗的效果。有效抗菌治疗方案需能够保证抗菌药物在机体的感染灶中达到足以杀灭或抑制病原菌的有效浓度并维持一定的时间,能够清除感染灶内的病原菌以实现治愈感染的目的,能够遏制细菌耐药性的产生,并能够同时尽可能降低抗菌药物对机体产生的不良反应。抗菌药物临床试验必须体现抗菌药物的特点,疗效评价必须同时评价杀灭或清除病原菌的微生物学疗效和临床疗效。
本指导原则所涉及的抗菌药物指具有杀菌或抑菌活性,主要供全身应用的抗菌药物。
(二)抗菌药物PK/PD研究意义及分类
抗菌药物药物代谢动力学(pharmacokinetics,PK,以下简称药代动力学)研究可定量描述其在机体血液循环、感染部位体液或组织中的浓度随时间变化的规律,抗菌药物药效学(pharmacodynamics,PD)研究反映其抑制或杀灭病原菌活性的高低。PK/PD研究将药物浓度与时间、抗菌作用结合起来,阐明抗菌药物在特定剂量/浓度和特定给药方案下抑菌或杀菌效果的时间过程。
抗菌药物PK/PD研究已被广泛应用于抗菌新药研发全过程。以抗菌药物体外PK/PD模型、动物感染模型和临床药代动力学研究包括经典PK和群体药代动力学(Population Pharmacokinetics,PPK)结合PD研究为基础,通过蒙特卡洛(Monte Carlo Simulation,MCS)等方法的基于模型模拟的药物研发(Model BasedDrug Development,MBDD)手段,为抗菌药物各期临床试验给药方案的制定、抗菌药物群体量效关系的探索、特殊患者群体和特定患者个体给药方案的调整等提供支持性数据;可为抗菌药物对各目标病原菌的药敏折点(susceptibilitybreakpoint)的制定提供PK/PD界值(pharmacokinetic/pharmacodynamic cutoff,PK/PD cutoff),并在剂型变化、新适应证增加、新适用人群、上市后给药方案优化以及药品审评审批和监管决策等方面发挥重要作用。
根据PK/PD理论一般将抗菌药物分为浓度依赖性(concentration-dependent)药物和时间依赖性(time-dependent)药物两大类。浓度依赖性抗菌药物杀菌效果与其药物浓度相关,浓度越高,则杀菌效果愈强。其主要的PK/PD指数为游离(以f表示游离药物的百分率)药物的血药峰浓度(fCmax)与最低抑菌浓度(minimal inhibitoryconcentration,MIC)的比值(fCmax/MIC)以及游离药物的药时曲线下面积(fAUC0-24)与MIC的比值(fAUC0-24/MIC)。代表药物如氨基糖苷类、喹诺酮类和硝基咪唑类。时间依赖性抗菌药物的游离药物浓度在对病原菌的MIC的4—8倍内,杀菌效果与浓度相关,但超过该浓度范围后,杀菌速率达饱和状态,其杀菌效果与药物浓度超过病原菌MIC时间的长短有关,则主要的PK/PD指数为游离的药物浓度高于MIC的时间占给药间期的百分比(%fT>MIC),代表药物如β-内酰胺类等。此类药物中某些抗菌药抗生素后效应(postantibiotics effects, PAE)无或短,如果PAE较长,则主要PK/PD指数为fAUC0-24/MIC,代表药物如糖肽类等。必须指出PK/PD分类并非绝对的和固定不变。
(三)依据PK/PD研究确定给药方案的基本原则
在抗菌药物剂量选择中,基本原则是首先考虑在该给药方案下保证患者安全性和耐受性,然后根据PK/PD研究选择取得最佳的临床和微生物疗效的剂量,且能有效防止细菌耐药性产生。优化给药方案一般策略为:对于浓度依赖性抗菌药物可通过减少每日给药次数或单次给药,使fCmax/MIC和fAUC0-24/MIC值达较高水平;对于时间依赖性无PAE的抗菌药物则日剂量分多次给药或延长输注时间(静脉制剂),使fT>MIC的时间延长;对于时间依赖性长PAE的抗菌药,一般在日剂量相同下,减少给药次数,使fAUC0-24/MIC值足够高。
(四)抗菌药物PK/PD研究的特点
抗菌药物PK/PD研究贯穿于抗菌药物研发的各个阶段,以支持抗菌新药有效性和安全性的确切评价。该研究随着药物研发的进程而逐渐完善,可以分为非临床阶段的PK/PD研究和临床阶段的PK/PD研究两部分。基于动物和人体感染的致病菌为同一类型,作用机制也一致的特点,因此抗菌药物体外药效学结合体外PK/PD研究和动物PK/PD研究等非临床PK/PD研究可间接反映抗菌药物进入机体后在感染病灶内达到抑菌或杀菌效果的动态过程,预测药物在人体内的杀菌和抑菌效果,此对临床PK/PD研究及给药方案制定具有重要参考价值,临床PK/PD研究是决定临床给药方案的制定包括剂量的优化、亚组剂量选择等重要依据。
(五)抗菌药物PK/PD研究策略
为了充分体现抗菌药物PK/PD研究的价值,缩短研发周期,降低研发风险,其研究策略必须清晰,整体研究计划和方案必须详细,结果分析及决策必须及时。
从整体上考虑,其研究策略包括但不限于如下内容:
(1)确定PK/PD指数
此部分研究主要在非临床阶段的体外或/和体内动物PK/PD研究中完成。临床PK/PD研究加以确认。
(2)确定PDT
在非临床阶段,通过体外或/和动物PK/PD研究获得抑菌或杀菌效果所需PK/PD靶值,又称药效学靶值(PDT);在临床阶段,结合临床的适应证和适用人群情况,进一步完善。
(3)同步开展抗菌药物对主要目标病原菌的MIC分布研究
(4)确定PTA
依据早期临床获得的药代动力学数据,如健康受试者的PK数据或目标适应证患者PK数据,结合抗菌药物对目标适应症病原菌不同菌种MIC分布,采用模拟和诸如蒙特卡洛仿真(Monte Carlo simulation,MCS)等方法,拟定或确定抗菌药物不同给药方案达到PDT的几率,据此筛选和评价不同给药方案等(包括剂量、间期和频率)。
(5)推荐确证性临床试验的给药方案
如抗菌药物对目标适应证主要病原菌的达标概率(一般在90%以上)满足要求时,所制订的给药方案则可推荐为确证性临床试验的给药方案。该部分研究应在确证性临床试验前完成。
(6)暴露-效应关系的优化、分析和应用
这部分研究可以在确证性临床研究结束后综合分析时确定,并需要在后续的Ⅳ期临床试验中继续完善。
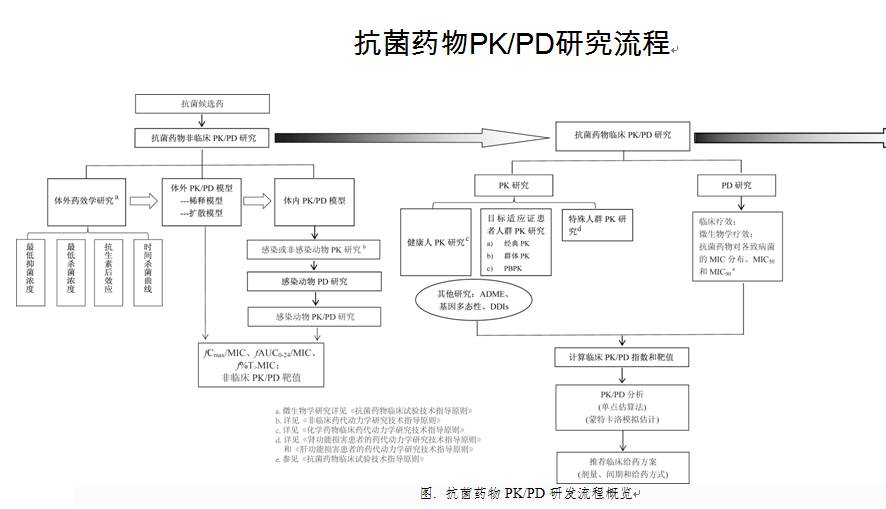
研究流程图见附件。
(六)本指导原则的目的及应用范围
制定本指导原则的目的是建立抗菌药物PK/PD研究技术规范,提升其研究质量,推进其在抗菌药物研发中的广泛应用,降低药物研发风险,为审评审批决策提供科学且充足的技术数据,为未满足的临床需要提供有力武器,保障科学用药。
本指导原则主要用于指导和评价新抗菌药物PK/PD研究,包括但不限于抗菌药物给药方案的确定,以及上市后给药方案的优化等。
创新抗真菌药物等临床试验也可参照。
二、非临床阶段的PK/PD研究
非临床阶段的PK/PD研究包括了体外研究和体内研究两部分,前者主要为体外药效学研究和体外PK/PD研究,后者主要为动物PK研究、感染动物PD研究和感染动物PK/PD研究。其目的是阐明抗菌药物的药效学特性,确定PK/PD指数和非临床PK/PD靶值。
(一)体外研究
1. 体外药效学研究
申报新药注册有关的微生物学研究详见《抗菌药物临床试验技术指导原则》等,此部分仅涉及PK/PD研究中的PD研究主要参数、技术方法和要求及结果描述。
1.1 最低抑菌浓度(MIC)
应根据抗菌药物自身的特点及未来可能拟定的临床适应证选择受试菌株,适当加大未来适应证中可能涵盖的病原菌研究。考虑到病原菌存在地区差异以及随时间的耐药性变迁等特性,入选的受试菌应满足以下要求:
(1)代表性:选择的受试菌应尽可能来自未来可能的适应证菌株,并尽量体现其野生株的特点。
(2)区域性:至少有3个区域的受试菌株进行汇总分析。
(3)近期流行:受试菌一般应选择近2—3年的临床分离菌株,以反映流行病原菌的敏感性和天然耐药特点。一些收集困难的菌种可考虑5年内的临床分离菌株。
应根据国际公认方法对各种受试菌进行分类和MIC测定。测定结果描述包括MIC范围、MIC50和MIC90、MIC众数等。
1.2 最低杀菌浓度(MBC)
根据抗菌药物对目标病原菌的MIC测定结果,选择具有药效学特点的细菌进行最低杀菌浓度(minimal bactericidal concentration, MBC)测定。每种细菌应包含不同MIC值的菌株。MBC测定根据国际公认方法,测定结果描述需包括MBC范围、MBC50、MBC90、MIC50/MBC50和MIC90/MBC90等重要参数。
1.3 抗生素后效应(PAE)
根据抗菌药物MIC和MBC结果,选择符合PAE测定要求的受试菌株,应覆盖拟定临床适应证的主要菌种。每种细菌应包含敏感株、有特殊耐药特征菌株以及同种细菌的质控菌株。结果描述需包括细菌生长和杀菌或抑菌曲线图以及PAE值。
1.4 时间杀菌曲线
时间杀菌曲线(time-kill curve)通常指静态杀菌曲线,是指固定一系列抗菌药物的浓度,观察药物对受试菌的杀菌活性以及杀菌速率随浓度的变化过程。
受试菌一般应覆盖拟定临床适应证的主要病原菌种,每种细菌应至少包含不同MIC值的菌株(包含野生型分布的高MIC值端菌株)及质控菌株。杀菌速率通过初始时间段菌落计数对数变化值与时间差的比值得到。结果描述需包括细菌菌落计数—时间图(即杀菌曲线图)、细菌杀菌速率—药物浓度曲线图和菌落计数—药物浓度对数量效曲线等。根据图示和反映量效关系的药效学模型(如Emax模型)等,分析该抗菌药的PK/PD特性属浓度依赖性抑或时间依赖性。药效学参数包括Emax、EC50、γ(Hill系数,反映曲线陡度)和模型拟合相关指数(R2)等。
2. 体外PK/PD模型
抗菌药物体外PK/PD模型(in vitro PK/PD model)是一种借助体外装置模拟抗菌药物在机体内药物浓度随时间变化(药代动力学过程)中抑制或杀灭细菌(药效学)动态过程,描述机体用药后抗菌药作用、细菌生长(或死亡)与时间的定量关系,也可称为体外动态杀菌模型,此模型可用于抗菌药物体外PK/PD指数及靶值的制定、给药方案(给药剂量、给药间隔)的筛选,尤其适用于基于细菌耐药机制的抗菌药PK/PD研究。
抗菌药体外PK/PD模型主要包括稀释模型和扩散模型,常用的扩散模型为中空纤维感染模型(hollow fiber infection model,HFIM)。
应根据抗菌药对目标病原菌体外药效学研究结果,选择拟订临床适应证的主要目标病原菌开展体外PK/PD研究。处于非临床研究阶段的药物可采用动物PK数据进行体外药时曲线模拟,但此数据仅供参考。一般在获得了健康受试者或患者PK数据后,结合这些数据做进一步的模拟。
在体外PK/PD模型中模拟抗菌药不同给药方案下药时曲线,其Cmax、AUC和t1/2等PK参数,结合该药对受试菌的MIC值,建立3个PK/PD指数fCmax/MIC、fAUC0-24/MIC和f%T>MIC与其药效学参数(细菌菌落计数变化值,ΔLog10CFU)的药效学模型(如SigmoidalEmax模型。根据拟合度大小选择代表该抗菌药的最佳体外PK/PD指数。将细菌菌落计数对数降低不同单位时(ΔLog10CFU取值0、-1或-2,分别对应细菌净生长为零、细菌菌落计数降低至1/10和1/100)fCmax/MIC、fAUC0-24/MIC或f%T>MIC的对应值作为PK/PD靶值(PDT)。结果描述中需包括不同抗菌药物给药方案下细菌菌落计数—时间图(即杀菌曲线图)、细菌菌落计数降低值—PK/PD指数(fCmax/MIC、fAUC0-24/MIC或f%T>MIC)图(即相关性分析图),并对体外PK/PD靶值(PDT)作描述性统计分析。此外,尚可采用该模型评价耐药细菌出现的情况,并对PK/PD靶值确立作相应分析。
(二)体内研究
动物感染模型(in vivo PK/PD model)可用于研究各种抗菌药物的不同给药方案进入感染或非感染动物体内PK特点、抑菌或杀菌效果(细菌菌落计数降低或动物存活率/死亡率)及治疗时间(疗程)的长短,据此获得的动物PK/PD指数及靶值,其与临床研究结果有较好的一致性,对临床和微生物学疗效的预测性优于体外PK/PD模型。现有动物感染模型如大腿感染、肺炎、心内膜炎、尿路感染、腹腔感染和全身感染模型等已用于治疗细菌性肺炎、血液感染、尿路感染、皮肤软组织感染和复杂性腹腔感染等的抗菌新药研发中。
动物PK/PD模型一般采用的感染动物为小鼠、大鼠等,一般同时在免疫缺陷感染小鼠和免疫正常感染小鼠体内评价抗菌药物体内活性。一般通过腹腔注射环磷酰胺的方法诱导嗜中性粒细胞减少以消除免疫状态对结果的干扰。常用免疫缺陷鼠大腿感染模型和免疫缺陷鼠肺炎模型等,其药效判断指标明确(组织中细菌菌落计数变化值),该方法重复性好和简单方便。有条件单位可同时采用微透析技术,动态测定小鼠/大鼠腿部感染、肺部感染组织中药物浓度,评价血液及靶组织中的PK/PD特性。可根据抗菌药自身特点及拟定临床适应证,选择相应血流感染模型或尿路感染模型等其他模型。
抗菌药物剂量的选择、PK参数的准确测定和药效学指标的设定等,对构建感染动物PK/PD模型成功与否至关重要。此部分仅以涉及感染动物PK/PD研究中技术方法和要求及结果进行描述。
1. 动物PK研究
动物PK研究内容、要求及结果分析等详见国家食品药品监督管理总局(CFDA)发布的《非临床药代动力学研究技术指导原则》。一般在非感染动物模型中测定抗菌药在动物体内不同时间点血浆(血清)浓度,计算抗菌药物不同剂量、不同给药频率(单次或多次)给药后动物的PK参数。必要时研究感染动物与正常动物PK的差异。如果动物使用过免疫抑制剂环磷酰胺,应研究环磷酰胺对动物PK影响,对于某些动物组织或部位如中枢神经系统、肺和皮肤中药物浓度明显不同于血浆/血清中的浓度,除血浆中药物浓度外,还应提供该感染组织或部位中药物浓度数据和组织体液穿透率等。
2. 感染动物PD研究
以小鼠腿部感染模型为例,在免疫缺陷或正常小鼠双侧大腿注射对数生长期的细菌,设立治疗组与阳性对照组(未给予药物治疗者),抗菌药物不同剂量、不同给药频率给予治疗组小鼠后,测定给药后不同时间感染动物局部组织或血液中细菌菌落计数,感染动物存活率/死亡率及存活天数等药效学指标。每种动物应包含不同MIC值受试细菌感染的动物。
必要时,进行动物体内PAE研究,观察动物体内抗菌药物全部消除后(即测不到动物体内药物浓度)细菌恢复对数生长的延迟时间,此有助于评价动物PK/PD特性。一般动物体内的PAE比体外PAE长,此可能与体内有亚MIC、血清因子等使细菌生长较缓慢所致。结果描述需包括感染动物体内细菌生长曲线、杀菌曲线以及体内PAE值。
(三)感染动物PK/PD研究
该研究可定量描述抗菌药物不同给药方案下感染动物血中药物浓度随时间变化以及与动物体内细菌菌落(CFU)计数变化、动物存活率或死亡率之间的关系,建立PK/PD指数(fCmax/MIC、fAUC0-24/MIC和%fT>MIC)与其感染动物组织/体液中细菌菌落数对数差值(ΔLog10CFU)的药效学模型,动物PK/PD靶值的分析过程参见体外PK/PD模型部分。亦可建立PK/PD指数与动物生存率/死亡率的药效学模型,获得反映治疗效果的最佳PK/PD指数及靶值。对研究株数的要求,应覆盖主要的目标适应证细菌,一般需要3—5株,且部分菌株的MIC应在其野生型分布的上端。感染动物模型可用于筛选抗菌药物不同给药方案(剂量、间隔、治疗天数)下取得的微生物学疗效(感染血液/组织中细菌菌落数降低)和治疗效果(动物存活率),推荐预期人体内最大杀菌效果和临床疗效的给药方案,为I期临床试验给药剂量范围确定、II、III期临床试验有效剂量的选择提供参考。结果描述参见体外PK/PD模型部分。
三、临床阶段的PK/PD研究
抗菌药物获准进入临床试验后,在制定各期临床试验方案时,需在临床各期中开展PK/PD研究,对临床试验给药方案的制定提供数据资料,支持抗菌药物有效性和安全性确切评价。
(一)PK研究
1. 健康人群PK研究
抗菌药物在健康受试者中的药代动力学研究是新药临床研究中必不可少的部分。其研究内容、要求及结果分析等详见《化学药物临床药代动力学研究技术指导原则》。该研究获得的PK研究数据可同时用于后续的PK/PD研究。
2. 目标适应证患者人群PK研究
在目标适应证患者中开展PK研究可了解抗菌药物在目标适应证感染患者中药代动力学特征及其影响因素,对制定有效给药方案尤为重要。研究方法包括经典药代动力学研究和群体药代动力学(PPK)研究等。
2.1 经典药代动力学研究
抗菌药物在目标适应证受试者中的PK研究设计基本同健康受试者的PK试验,入选的目标适应证患者需病情稳定、无并发症和少有合并用药,一般为6—12例。以临床推荐的给药方案用药,一类抗菌药物宜采用探索性或确证性临床试验中的给药方案。可单剂量给药或多剂量给药达到稳态后在一个给药间期内多点采集患者的血样,用于经典PK参数的计算。如果该抗菌药属非线性PK特征或前期的健康受试者PK结果显示多剂量给药后呈积蓄,有必要在感染患者中开展多种剂量给药后在不同给药间隔内采集血样,进行PK计算和统计分析。结果描述需包括PK参数。统计分析时需包括与健康人体PK参数比较。如果样本量足够时,需按性别、体重及年龄等分组后比较PK参数的差异性。
2.2群体药代动力学研究
抗菌药物注册临床试验建议同步开展PPK研究,通过稀疏点采样方法采集感染患者不同时间点的血样,收集患者生理(年龄、性别、体重等)、病理(肝、肾功能损害和其他疾病状态等)和合并用药等影响患者体内PK参数的各种数据(固定效应因素或协变量),建立PPK模型,定量描述抗菌药在患者体内PK过程,以及患者群体间存在的PK差异,确定主要影响PK的因素、对制定各感染患者群体的给药方案非常具有价值,尤其对一类抗菌新药探索性和确证性临床研究中各期给药方案的制定尤为重要。抗菌药物群体药代动力学研究内容、要求及结果分析等详见《群体药代动力学研究技术指导原则》。
PPK研究可在临床试验一部分患者中开展,更推荐全体入组患者,后者可以获得更多的研究信息。
3. 特殊人群PK研究
抗菌药物在特殊人群如肝、肾功能减退者的药代动力学研究内容、要求及结果分析详见《肾功能损害患者的药代动力学研究技术指导原则》和《肝功能损害患者的药代动力学研究技术指导原则》。
需特别强调,抗菌药物在肝功能或肾功能损害患者的药代动力学研究不同于其他药物,伴有研究药物治疗适应证的肝、肾功能受损的感染患者不宜纳入研究,仅选择伴有肝功能或肾功能损害的受试者开展此类研究。
对于一类抗菌新药,在健康受试者PK研究完成后,根据其体内代谢、排泄特点,应尽早开展此类研究,以便为确证性临床研究中入排标准和给药方案的制定提供依据。
尽管在抗菌药物的探索性和确证性临床研究中开展了PPK研究,但受限于入排标准,因此不可能纳入重度肝、肾功能减退患者,因此尚需单独开展此类特殊群体的PK研究。
4. 其他
要全面了解抗菌药物在人体内吸收、分布、代谢和排泄等临床药理学过程,需要进行以下研究:
4.1 组织分布及穿透性研究
抗菌药物入血后,与血浆蛋白呈可逆性结合,一般认为从毛细血管中游离出来的抗菌药物到达感染部位的细胞外液才能发挥抑菌或杀菌作用,因此,研究中需先获得该药的血浆蛋白结合率。进行PK/PD分析时,一般采用游离抗菌药物浓度或暴露量进行计算,以游离药物百分率(1-血浆蛋白结合率)与血药浓度及暴露量乘积(fCmax/MIC和fAUC0-24/MIC)来反映感染组织体液PK/PD与疗效相关性,此更具指导意义。
分析抗菌药物在组织体液的总浓度-时间曲线数据和/或组织体液中的游离浓度-时间曲线数据,并与血浆(血清)药时曲线关联,对指导给药剂量的选择有重要意义。较重要的组织体液浓度数据包括:①治疗尿路感染时,需提供抗菌药物尿药浓度及尿排出数据;②治疗肺炎的药物需提供抗菌药在上皮细胞衬液(ELF)中的浓度数据;③治疗中枢神经系统感染药物需提供在脑脊液(CSF)中的药物浓度。通常以非感染患者为研究对象进行组织穿透性评价,鼓励从目标适应证患者中获得组织体液药物浓度数据。
4.2 代谢产物活性研究
抗菌药物在体内通过肝脏或肠道等器官代谢后,代谢物可保持原有抗菌活性,或抗菌活性减弱或消失。如果在健康受试者PK研究中,发现代谢产物暴露量(AUCmetabolite)占母药暴露量(AUCparent)10%以上时,需对该代谢产物进行体外药效学研究,包括该代谢产物对目标适应证主要病原菌MIC50、MIC90和MIC范围等,从而了解代谢物的抗菌谱和抗菌活性。此项研究需在该抗菌药确证性临床试验开始前完成。当代谢产物PK参数个体间差异较大,半衰期较长时需关注其是否有抗菌活性。
4.3 基因多态性
对于某些抗菌药物,由于基因多态性导致其体内PK变化,在PK/PD研究时需加以分析和评估。
4.4 药物相互作用研究
对于某些药物,由于药物间相互作用导致PK变化,在PK/PD研究时需加以分析和评估。抗菌药物相互作用研究内容、要求及结果分析等详见《药物体内代谢和药物相互作用研究指导原则》。
(二)PD研究
抗菌药物的探索性和确证性临床试验中,获得其在目标适应证患者的临床疗效和微生物学疗效,测定临床分离致病菌MIC,其结果可反映该试验药对临床目标病原菌等临床致病菌的抗菌活性,与临床疗效和微生物疗效进行相关分析。
抗菌药物临床和微生物学疗效的指标和判断标准参见《抗菌药物临床试验技术指导原则》。
(三)临床PK/PD关系建立
动物或体外PK/PD关系研究尚不能完全和准确反映临床PK/PD关系,在获得了临床阶段的PK和PD数据后,应开展临床PK/PD关系建立研究,其研究内容包括临床PK/PD靶值的制定和临床各期给药方案的制定,其技术方法、要求及结果描述如下。
1. 临床PK/PD指数及靶值
在目标适应证患者群体中,采用稀疏点采样方法,开展抗菌药物在感染患者中的PPK研究,建立PPK模型,采用Bayesian等获取感染患者个体PK参数,结合该患者自身感染病原菌MIC值,计算该患者的PK/PD指数值(fCmax/MIC、fAUC0-24/MIC或f%T>MIC),并与该患者临床疗效(治愈或失败)、微生物学疗效(病原菌清除或未清除)或综合疗效(临床疗效和微生物疗效综合,治愈或失败)建立药效学模型(如Logistic回归模型),据此从临床PK/PD角度确定该药的PK/PD指数,同时确定其临床PK/PD靶值。一般而言,临床治愈率、细菌清除率达到90%时的PK/PD值即为体内达到最大杀菌效果临床PK/PD靶值。结果描述需包括临床治愈率、细菌清除率或综合疗效率与PK/PD指数(fCmax/MIC、fAUC0-24/MIC和f%T>MIC)相关性图。临床PK/PD靶值平均值、标准偏差和95%可信区间等。
根据临床PK/PD靶值筛选和优化抗菌药物给药方案最具临床意义,但需要注意的是:在一些抗菌药物临床研究中,由于细菌培养阳性率不高,患者同时获取PK和PD数据较为困难;临床研究中分离得到的细菌种类和数量缺乏代表性等因素,导致PK/PD指数和其疗效间的定量关系较难建立,不易获得临床PK/PD靶值,此时可参考非临床PK/PD分析及结果。
在这一阶段的PK/PD关系建立中,由于还可以获得临床疗效指标和安全性指标,可以构建范围更宽的量效关系模型,用于指导临床给药方案的选择。
2. 确定临床给药方案的方法
以临床研究中获得的PK/PD靶值为目标,通过采用MCS等方法筛选给药方案。其具体内容为通过比较抗菌药物不同给药方案(给药剂量、间隔及给药方式)所得PK/PD指数达到靶值的概率(一般为大于等于90%;如果达到90%概率所需剂量的耐受性较差且需要抗菌药治疗感染疾病时,或者感染病严重程度较低,且MIC值达到90%概率所需MIC值范围上限的菌株极少时,概率低于90%也可以接受),确定临床给药方案。由于临床PK/PD靶值在I期临床研究、探索性临床研究,甚至是确证性临床研究中很难获得,建议前期采用动物或体外PK/PD靶值或参考同类药物的靶值予以假设。MCS计算的前提是必须有PK/PD靶值,由于不可能对目标适应证所有病原菌进行PK/PD研究,可采用单点估计法进行不同临床给药方案间的比较。
2.1 单点估计法
PK参数来自健康受试者或患者(经典PK或PPK研究中获得),PD参数来自体外药效学研究中MIC结果(MIC50和MIC90),或来自于探索性和确证性临床研究中分离自患者致病菌的MIC结果(MIC50和MIC90),后者更具临床参考价值。如果某些细菌株数少于10株,无法计算MIC50和MIC90值,则无需进行单点估计法分析。单点估计法计算抗菌药物不同给药方案下PK/PD指数fCmax/MIC、fAUC0-24/MIC或f%T>MIC数值,其中MIC指MIC50和MIC90,评价抗菌药物不同给药方案下对不同菌种的不同细菌的PK/PD值的大小,综合分析后建议临床给药方案。
该研究方法缺陷:PK和PD数据均为单点估计值,未考虑PK参数个体间和个体内变异以及细菌MIC分布情况。
2.2 蒙特卡洛模拟
蒙特卡洛模拟(MCS)指采用来自健康受试者或/和患者PK或/和PPK数据(平均值及变异值)进行模拟,产生模拟数据。模拟时,需要按照数据的实际分布,通过相应的参数进行模拟。为了获取可信的结果,以及95%的置信区间,需要进行多次模拟平均值及变异值产生模拟数据)。PD参数通常为体外药效学研究中MIC结果(MIC分布),或临床试验中分离自患者致病菌的MIC结果(MIC分布)产生的模拟数据。根据其PK/PD特点,计算fCmax/MIC、fAUC0-24/MIC或f%T>MIC,以非临床或临床PK/PD靶值为目标,计算抗菌药物不同给药方案对某一细菌PK/PD指数达到该靶值的累积响应百分率(cumulative fraction of response,CFR),以及不同MIC值下对某一细菌达到PK/PD靶值的达标概率(probability of targetattainment,PTA)。一般而言,抗菌药物给药方案对某一细菌CFR值高于90%时,提示该给药方案对该细菌具有最大杀菌效果,为优选方案。当给药方案在某一MIC值下的PTA值高于90%,且该MIC值位于野生型菌株MIC分布范围上端(包括MIC90和/或流行病学界值)时,认为此给药方案为有效治疗方案。该MIC值也是抗菌药物对该细菌的PK/PD界值(PK/PD cutoff)。根据PTA和CFR结果:①确定达到最佳临床和细菌学疗效时抗菌药物的给药方案,包括给药剂量、间隔和给药方式(如注射液为间断或连续静脉输注等);②抗菌药物对各目标病原菌最大杀菌效果,供临床制定目标适应证所覆盖目标病原菌种类参考;③PK/PD界值的确定,为抗菌药物对目标病原菌药敏折点的制定提供依据。
进行PTA分析时,可按抗菌药MIC值和PK/PD靶值(ΔLog10CFU取值0、-1或-2)列出分析结果。进行CFR分析时,建议按病原菌种属和PK/PD靶值列出分析结果。如果分析目的是使临床疗效和微生物学疗效有效率与疾病治疗最佳条件下的有效率相当,可根据以下选择靶值:①感染可能危及生命时(有较高的病原菌负荷且自发缓解率较低,如医院获得性肺炎或呼吸机相关肺炎),通常选择菌落计数至少降低10倍(ΔLog10CFU≥1)所需的PK/PD指数作为靶值;②感染有较轻的病原菌负荷,或者可通过抗菌治疗联合其他干预措施治疗时(如急性细菌性皮肤和皮肤结构感染以及腹腔感染,这些感染常采用手术治疗),选择至少抑制细菌生长(ΔLog10CFU≥0)所需的PK/PD指数作为靶值。
该研究方法缺陷:必须有PK/PD靶值,但临床试验前期较难获得,参照同类药物相关PK/PD靶值予以PK/PD分析,可能对给药方案评价造成偏倚。因此建议将上述两种PK/PD分析方法结合进行抗菌药物不同给药方案评价。
四、PK/PD研究的应用
(一)PK/PD研究应用于研发决策
目前可以通过非临床PK研究结果预测人体PK参数,如体外模型、种属间比放(scaling)、基于生理模型的药代动力学(PBPK)研究等,并且有成功的案例。
研究单位可以充分利用这些手段,结合非临床研究获得PK/PD靶值,预测人体中可能的有效剂量,分析候选抗菌化合物的开发价值,以决定是否继续研发。
(二)PK/PD研究应用于I期临床试验
1. 初步预测临床给药方案,为继续研究提供决策
根据I期临床试验健康受试者在不同给药剂量下抗菌新药单剂或多剂给药后PK数据,结合体外药效学的MIC结果,以动物或体外PK/PD靶值(也可参考同类药的PK/PD靶值)作为临床剂量选择的参考依据,采用单点计算法或MCS方法计算在人体达到同样PK/PD靶值时的临床用药剂量。如果I期临床耐受性试验结果显示,达到该剂量时受试者可能存在耐受性问题,则意味着该候选抗菌新药进入探索性临床研究阶段存在风险,申办者需要慎重决策进一步研究。
2. 指导探索性临床试验的给药方案制定
抗菌新药I期临床PK/PD分析结果显示探索性临床试验预测给药方案在健康受试者中耐受且安全性良好时,应在I期临床试验报告中,报告PK/PD分析方法和结果,并提出探索性临床研究中目标适应证和给药方案的建议。
(三)PK/PD研究应用于探索性临床试验
探索性临床试验目的是探索抗菌药物适应证和最佳给药方案。在该研究阶段,如同时还开展PK研究(经典PK或PPK),可以获得目标适应证患者的PK参数和其变异,以及该研究药物对标适应证病原菌的MIC数据,此可用于构建准确的临床PK/PD关系,指导给药方案的选择。
抗菌新药探索性临床试验(II期)中,建议同时开展目标适应证患者经典PK研究和PPK研究。根据I期临床试验的结果,制定患者的PK及PPK采样点,需兼顾抗菌药物在患者的吸收、分布、代谢和消除各相,同时需考虑临床可操作性。
探索性临床试验为剂量探索,入组的病例数有限,因此临床试验结束后,较难建立PPK模型和PK/PD评价。建议申办方或研究者根据患者经典PK数据,结合研究药物对目标适应证病原菌的MIC数据,采用单点估计法或MCS方法进行PK/PD分析并评价给药方案。建议将该研究结果与I期临床PK/PD研究结果进行比较,并分析与患者临床疗效和微生物学疗效的相关性。
如果抗菌新药研发的是经过充分评价的药物类别,且PK/PD研究较充分,如已有了充分的非临床PK/PD研究数据、并有代表性的目标适应证群体的PK数据等,则可以在经过充分论证后考虑豁免探索性阶段的临床试验。
抗菌新药探索性临床试验报告中,需报告患者经典PK研究结果或\和PPK、PK/PD分析方法和结果,并推荐确证性临床试验给药方案。
(四)PK/PD研究应用于确证性临床试验
在抗菌新药确证性临床试验中须进行PPK研究,稀疏采样点应基于已经完成的PK或PPK研究所获得的患者PPK特征,或文献资料进行设计,并考虑临床试验的实际情况,对探索性临床试验中PPK采样点进行相应调整,尽可能与患者实验室检查采血点一致,提高患者的依从性。在临床试验结束后,进行PPK建立及分析完善PK/PD关系,或暴露量/效应关系,用于制定不同亚群的给药方案、完善说明书等。
1. PPK模型的建立及应用
抗菌新药确证性临床试验完成后,如能判定可以整合此前的研究数据,则需将该研究药物在健康受试者和感染患者全程PK和PPK研究中所有血药浓度数据及人口统计学、临床和实验室检查数据整合,构建PPK数据集,建立PPK模型并予以验证,定量分析影响该药在人体内PK参数的因素,并按协变量分层后统计分析,比较各感染患者群体的生理和病理因素(如不同年龄、性别、体重或不同肝、肾功能情况等)对PK参数的影响,从而进一步说明患者处于不同生理和病理情况时,该抗菌药物确证性临床给药方案在人体内药物暴露量或药物消除等PK特性是否发生变化,以及是否需要对各患者人群的给药方案予以调整。
2. 暴露-效应关系的建立及应用
抗菌新药确证性临床试验结束后,测定抗菌新药对患者临床分离菌株的MIC。根据前述的“临床PK/PD指数及靶值”方法获得临床PK/PD靶值后,采用MCS等方法制定该抗菌新药在目标适应证患者中,基于该药对目标病原菌不同MIC值,以及不同生理和不同病理状态下或合并用药下,各患者群体达到最大杀菌效果的给药方案。此研究结果可用于注册上市临床给药方案的制定;药品说明书可列出有关健康受试者PK及目标适应证PK特征的描述,以及目标病原菌的PK/PD界值。
当抗菌新药临床试验疗效评价结果详实,抗菌药物PK和MIC数据足够多时,可以进行暴露-效应(Exposure-response, E-R)分析。需根据疗效变量及PK/PD指数的形式选择合适的统计学模型。如为临床疗效时(治愈或失败),常采用Logistic模型进行暴露-效应关系分析;疗效和PK/PD指数均为连续变量时,可采用Emax模型进行分析。除PK/PD指数外,如发现其他因素(如受试者年龄等)也能预测疗效时,需采用多变量分析评价每个因素对疗效产生的贡献。采用统计学模型拟合暴露-效应数据后应进行模型诊断,并评价模型预测性能。
暴露-效应关系分析可用于①寻找抗菌新药给药方案发挥疗效所允许的最高MIC值;②临床PK/PD靶值;③特定MIC值下效应产生的概率。
如果临床试验结果显示临床失败病例数少,或目标病原菌分离率低,无法建立PK/PD指数与临床疗效或微生物学疗效的定量关系,无法获得临床PK/PD靶值时,建议利用动物或体外PK/PD靶值,采用MCS等方法进行给药方案评价。此阶段不得参考同类药物PK/PD靶值进行相关的临床PK/PD分析。
抗菌新药确证性临床试验报告中,需报告PPK及PK/PD分析计划书和报告,报告中需全面描述PPK、PK/PD分析方法和结果,并推荐申请注册上市临床给药方案。
3. 特殊人群等的PK/PD研究
抗菌新药申请注册的临床试验通常有较严格的入排标准,无法入组一些特殊人群患者,对一些特殊人群如中度、重度肝肾功能减退者的PK特性尚不了解,无法提出特殊人群的给药方案推荐。探索性临床试验初步确定有效临床给药方案后,在确证性临床试验中,同期开展抗菌新药在特殊人群包括但不限于肝功能和/或肾功能减退者、基因多态性、部分药物间的相互作用等PK研究。建议申办者在I期临床试验结束后,根据药物PK特点,尽早开展在特殊人群中PK研究。通过PK/PD分析制定特殊患者人群给药方案。这些临床研究报告应附在确证性临床试验报告中,其研究结果应纳入药品说明书。
(五)上市后研究
在注册上市后的大样本临床试验中,随着更广泛人群的加入,应继续开展抗菌药物PPK和PK/PD等相关研究。在大样本人群中观察药物浓度及暴露量与临床和微生物学疗效的相关性。临床试验结束后,应将上市前后所有人体药物浓度数据及人口统计学、临床和实验室数据合并,进行PPK及PK/PD分析,确认抗菌药物给药方案;进一步推荐特殊人群(如老年人)用药方案和合并用药情况下给药方案;完善药品说明书。
确证性临床试验中尚未完成的特殊人群、基因多态性和药物间的相互作用等研究必须在上市后临床研究中完成,研究结果载入药品说明书中。
新药注册上市后,申办者至少完成一项该抗菌新药在目标适应证患者中组织体液分布及穿透性研究,并通过感染部位PK/PD分析,对临床推荐给药方案进行评价和优化。如该药目标适应证为社区获得性肺炎,需开展抗菌药物在肺组织体液中PK/PD研究及给药方案评价。
(六)PK/PD在制定细菌敏感性折点中的应用
抗菌药物对目标病原菌药敏折点由流行病学界值(Epidemiological cut/off value,ECV)、非临床PK/PD界值(体外和动物PK/PD界值)和临床PK/PD界值三部分研究结果综合评估后制定。
根据抗菌药物临床研究中PPK及PK/PD研究结果制定PK/PD界值,供药敏折点制定时参考。
因抗菌新药I期临床试验和探索性临床试验尚处于剂量探索性阶段,并且PK及PPK研究受试者例数有限,一般在确证性临床试验中进行PK/PD界值的研究。根据I期、探索性和确证性临床试验等相关数据建立的PPK模型获得PK数据,结合非临床体外药效学数据和临床试验中目标病原菌MIC值及分布,以临床PK/PD靶值作为取得最佳临床疗效和微生物疗效靶标,采用MCS等方法计算在临床推荐给药方案下,对目标病原菌各MIC值下的PTA进行计算,一般以PTA≥90%时的MIC值作为PK/PD界值,用于确定药敏折点。如果无法获得临床PK/PD靶值,首选采用动物PK/PD靶值,其次为体外PK/PD靶值,依此方法获得的界值仅为初步的PK/PD折点,需在上市后临床研究中进一步进行验证。通过PPK及PK/PD获得PK/PD靶值,据此制定的PK/PD界值更能反映抗菌新药在临床推荐的给药方案下其临床和微生物学疗效的结局,对药敏折点的制定更具有价值。
(七)PK/PD在制订β-内酰胺酶抑制剂合剂给药方案中的应用
制定β-内酰胺类抗生素和β-内酰胺酶抑制剂联合(以下简称“β-内酰胺酶抑制剂合剂”)给药方案时,需阐明β-内酰胺酶抑制剂对不同β-内酰胺酶有不同抑制活性,可通过以下研究评价抑制酶活性范围:①通过酶动力学试验综合评价抑制酶活性。②β-内酰胺类抗生素单用以及与不同浓度β-内酰胺酶抑制剂联用的体外研究,通过MIC测定和杀菌曲线实验评估抗菌活性;所选细菌包括天然产β-内酰胺酶的菌株,或者通过基因工程修饰后产β-内酰胺酶的菌株。
应针对每个β-内酰胺类抗生素和β-内酰胺酶抑制剂建立预测疗效的最佳PK/PD指数。在建立PK/PD指数的过程中,开展的研究应包括非临床感染模型。其中,β-内酰胺类抗生素和β-内酰胺酶抑制剂应模拟临床给药(按给药间隔间断给药或持续滴注)后的体内过程,并记录Cmax、AUC0-24和浓度高于域值(抑制β-内酰胺酶活性最低浓度)时间占给药间隔百分比(%T>threshold)等PK参数;然后结合MIC数据进行PK/PD分析(如计算PTA),最终确定给药方案。
计算β-内酰胺酶抑制剂合剂达标概率时,需先分别建立β-内酰胺类抗生素和β-内酰胺酶抑制剂的PPK模型,分析β-内酰胺类抗生素和β-内酰胺酶抑制剂在血浆中暴露程度的变异,以及两者同时给药可能引起的PK相互作用等因素后进行模拟计算,获得达标概率值。
对于主要经肾排泄的β-内酰胺类抗生素和β-内酰胺酶抑制剂,应通过模拟评估肾功能减退时给药剂量是否需要调整,尤其适用于β-内酰胺类抗生素总清除率(或肾清除率)和β-内酰胺酶抑制剂不同时。当β-内酰胺类抗生素和β-内酰胺酶抑制剂在临床上按固定配比给药时,如果PK/PD分析结果显示β-内酰胺类抗生素剂量调整方案并不适用于β-内酰胺酶抑制剂,提示肾功能减退患者避免使用该合剂。
通过临床试验评价β-内酰胺酶抑制剂合剂新给药方案时(以已知给药方案为参照),应注重选择对β-内酰胺类抗生素不敏感但对β-内酰胺酶抑制剂合剂敏感的产β-内酰胺酶病原菌感染的患者人群进行评价。PK/PD分析结果可用于评价患者应用β-内酰胺酶抑制剂合剂后受益与风险之间的关系。
五、PK/PD研究注意事项
(一)PK/PD研究局限性
抗菌药物PK/PD研究对给药方案的制定、优化和药敏折点制定等具有重要意义,但也存在着一定的缺陷和局限性,如不能预测药物起效的快慢、疗程等。在PK/PD研究中需要注意:
1. PK数据代表性及可靠性
抗菌新药注册上市前的PK数据来自于健康人体和感染患者,样本量有限,此可能导致注册上市前PK研究结果的群体代表性不够。
研究者在采用人体给药后不同时间下药物浓度值及蛋白结合率等数据计算PK参数时,必须确保该类数据的完整性、规范性和一致性,确保PK计算方法和过程准确无误,以及生物样品分析测试结果能够准确反映其体内的变化过程,详细地分析和评估多种影响因素对分析测试结果带来的干扰,最终确认PK数据的代表性和准确性。否则,基于该PK结果的所有PK/PD分析均无意义。
2. PD数据代表性及可靠性
由于受感染患者临床分离株的数量、质量以及病原菌地区分布影响,PD指标具有复杂性,且细菌耐药性变迁等进一步增加PD数据的不确定性,从而对PK/PD分析及临床推荐给药方案的确定带来一定影响。通过非临床PD数据和临床研究中PD数据的积累来降低此不确定性。
3. PK/PD分析数据及其代表性
一般而言,抗菌药物在感染组织中PK/PD分析结果与临床疗效结果相关性最好,但多数情况下难以获得组织PK数据,通常以人体循环PK数据来代替组织PK数据。考虑到细菌性感染常发生在细胞外液,抗菌药在细胞外液的游离浓度一般与血液循环中的游离浓度具一致性,因而用抗菌药物在血浆的PK参数反映其在感染病灶组织中的暴露量和维持时间是可行的。但有时该假设不成立,组织浓度和血浆浓度会存在显著差异,如肺部组织、中枢神经(脑脊液)和皮肤组织等,抗菌药物在组织和体液中的浓度可能显著高于或低于血浆浓度。此外,对于主要经过胆道、肾脏排泄的药物,其在胆汁或尿液的浓度也可能明显超过血药浓度,在此种情况下,以体循环中的PK参数代替组织中PK特性显然不恰当,需根据组织的PK参数对给药方案进行评价。因此建议申办方在抗菌药物临床试验中,尽早开展抗菌药物组织体液分布及穿透性研究,为体循环PK支持组织PK数据提供有利的证据。
此外,由于药物从体循环进入组织内经穿透过程,组织体液中药物峰、谷浓度均滞后于相应的血药浓度,因此基于血药浓度获得的Cmax/MIC值可能被估计过高,而%T>MIC值则估计过低,在制订给药方案时需综合考虑这些影响因素。
某些抗菌药物在体内可能存在多于一种活性形式,且活性产物之间的抗菌活性也不一致,此使PK/PD分析复杂化。在选择PK数据时,不仅要考虑主要活性产物的PK数据,对于活性代谢产物暴露量(AUC)高于母药暴露量10%者,均需进行PK/PD分析,并与母药进行综合PK/PD分析后评价给药方案。
一些PK/PD分析进行了复杂的建模和仿真操作,在分析过程中,需要对所构建的模型进行验证,只有符合预设验证标准的模型计算结果才有临床应用价值。
在PK/PD分析过程中,如果发现抗菌药物在体内的处置存在基因多态性时,需将基因多态性作为协变量加入到PPK及PK/PD分析中,为此类患者人群的给药方案制定提供参考。
进行某一抗菌药的PK/PD分析时,如果有同类已上市药,可将该药PK/PD分析结果作为参比,加以比较,除了验证受试药物计算过程的可靠性外,也为制定其合理临床给药方案提供支持性数据。
4. 机体免疫作用
一般在抗菌新药上市前的临床试验中,不会纳入免疫功能缺陷患者参加临床试验。建议申办方在非临床PK/PD研究中,通过对免疫正常和免疫缺陷感染动物的PK/PD研究,提出对免疫缺陷患者的给药方案建议。由于人体和动物存在差异性,鼓励在抗菌新药上市后临床试验中开展在免疫缺陷和免疫正常患者中随机对照临床试验。如果该药在感染动物中有充分和详实的实验数据和分析支持免疫功能缺陷给药方案的推荐,可考虑适当减少临床试验患者病例数。
5. 儿科人群的PK/PD研究
目前尚不能完全在抗菌药物上市前开展和/或完成儿科人群临床试验,建议申办方或研究者通过一些生理药动学模型(PBPK)和成人PPK模型构建,用基于成人的PK数据预测抗菌药在儿童体内的PK,并结合体外药效学数据做PK/PD分析,为儿童临床试验给药方案的拟定提供参考性建议。在该抗菌药上市后临床研究中,可采用PPK研究方法在儿科患者群体中获得PK数据,比较儿科人群和成人PK数据的差异性,并通过PK/PD分析后评价其给药方案的合理性,促进儿科人群用药的研发。需注意的是由于某些目标适应证中,成人和儿童感染致病菌敏感性会有所不同,建议在进行儿童PK/PD分析时,体外PD数据采用分离自儿童致病菌株的MIC结果。
6. 罕见病/少见菌的PK/PD研究
对于一些罕见致病菌引起的感染,如果无法开展针对该目标病原菌的临床试验,建议采用体外PK/PD研究方法筛选对该致病菌具有杀菌效果的给药方案,然后在感染动物PK/PD模型中予以验证,据此推荐给药方案。
(二)PK/PD研究报告格式及要求
进行PK/PD分析前,需要制定PK/PD研究方案及分析计划。PK/PD研究需按照方案及分析计划要求进行。PK/PD研究报告格式及要求如下。
1. 格式
本报告结构同其他报告的要求。区别点在于,PK/PD研究报告是随着药物研发进程逐步完善的,提交的PK/PD报告需体现这一点。I期临床试验中PK/PD分析可在I期研究报告中加入。探索性和确证性临床试验中PK/PD分析计划和研究报告需单独提交。
2. 数据来源
报告中须明确说明所采用的PK和PD数据来源。某些数据来自体外试验,动物或文献报道时,需注明出处。当某些参数(如蛋白结合率)有不同数值时,需要说明选择其中之一的理由及其合理性。
报告的附录中应提供PK和PD数据库。
3. 计算方法
报告中须详细说明PK/PD计算方法,例如采用单点估计法时,需说明PK参数和PD参数来源;采用蒙特卡洛模拟时,需说明PK参数和PD参数的分布假设、模拟次数,所采用的软件等,如果是计算%T>MIC还要说明计算原理。
根据PPK模型分析得到PK参数时,需详细说明PPK分析步骤及流程、试验数据的前处理方法、基础模型和协变量模型的建立方法、所用软件的类型和算法等。PPK模型包括模拟研究时,尚需描述模型的模拟方法、模拟受试者的基线数据来源等。
所有数据均须具可溯源性。报告的附录中应提供PPK和PK/PD计算程序代码。
4. 验证
PPK模型验证时,需提供验证的方法,应以表和图等形式充分说明所构建的PPK模型稳定可靠。
5. 结果和结论
应通过表格、图形和公式等详细展示分析结果,如病原菌MIC的分布特征图、PTA曲线、CFR数据表和药效指标对PK/PD指数的Logistic回归示意图等。列出PK/PD靶值的均值、标准差和95%置信区间。PPK结果中应包括研究人群的基线情况、PPK最终方程、PPK模型诊断图、模拟受试者的药时曲线等。建议按协变量分层进行不同群体间PK参数差异性的统计分析,对不同患者群体是否需要调整给药方案予以讨论,结论应根据PK/PD研究结果明确地推荐不同患者群体的具体临床给药方案。
6. 其他
PPK、PK/PD分析过程中,如果有漏检测数据或异常数据剔除,必须说明处理原则或剔除理由。有需要特殊说明之处,也必须在报告中加以解释。报告附件应详细列出所有引用药物研发的资料及参考文献。
六、名词解释
1. 药物代谢动力学(pharmacokinetics, PK,简称药代动力学):定量描述药物在机体内吸收(absorption)、分布(distribution)、代谢(metabolism)和排泄(excretion)的过程及药物浓度随时间动态变化的规律,可概括为ADME过程。反映ADME过程的主要PK参数包括:血药高峰浓度(Cmax)、达峰时间(Tmax)、药时曲线下面积(AUC)、表观分布容积(V)、药物清除率(CL)、表观分布容积和末端相消除半衰期(T1/2)等。
2. 群体药物代谢动力学(populationpharmacokinetics, PPK):指将经典的药代动力学模型与群体统计学模型(population statistical model)结合,研究药代动力学特性中存在的变异性,研究药物体内过程的群体规律、药代动力学参数的统计分布及其影响因素。
3. 药效学(pharmacodynamics,PD):指药物效应的大小随时间的变化过程。对抗菌药物而言,是指药物在体外或体内抑制病原菌生长和复制(抑菌)或致病原菌细胞死亡(杀菌)的作用。主要药效学参数包括抗菌药物对细菌的最低抑菌浓度(MIC)、最低杀菌浓度(MBC)、抗生素后效应(PAE)、亚抑菌浓度下的抗生素后效应(postantibiotic sub/MIC effect,PA/SME)和防突变浓度(mutationprevention concentration,MPC)等。
4. 最低抑菌浓度(minimum inhibitoryconcentration, MIC):体外抗菌药物敏感性试验中,抑制培养基内病原菌生长所需的最低药物浓度。
5. PK/PD指数(PK/PD index):药物暴露量(如药时曲线下面积)与参数(如最低抑菌浓度)相结合的定量指标。
6. PK/PD靶值(PK/PD target, PDT):效应指标达到预期水平时,所需的PK/PD指数值。
7. 蒙特卡洛模拟(Monte Carlo simulation,MCS):考察PK/PD指数在大量人群中分布规律的统计试验方法。首先根据PK参数和PD参数的分布特征进行随机抽样,然后将随机数值代入公式计算PK/PD指数,获知其分布规律,最终得到PK/PD指数达到靶值的概率。
8. 达标概率(probability of targetattainment, PTA):某MIC值下,通过模拟计算(如蒙特卡洛模拟)得到PK/PD指数达到PK/PD靶值的概率。
9. 累积响应百分率(cumulative fraction ofresponse, CFR):通过模拟计算(如蒙特卡洛模拟)得到PK/PD指数达到PK/PD靶值的概率。
10. PK/PD界值(PK/PD cutoff):达标概率超过90%时的MIC分布范围上限。
11. 杀菌曲线(time-kill curve):反映抗菌药物对细菌杀菌活性随时间变化的曲线。常指静态杀菌曲线,即固定一系列抗菌药物浓度,观察细菌与抗菌药物混合后的在不同时间点的菌落计数。以时间为横坐标,logCFU·mL-1为纵坐标绘制杀菌曲线。根据该曲线还可以计算杀菌速率,分析杀菌速率随浓度的变化。
12. 暴露-效应关系(exposure-responserelationship, E-R):药物在血浆/血液中的暴露量与治疗结果(如临床疗效)的关联。
13. 药敏折点(breakpoint):根据抗菌药物抑制细菌生长所需要的MIC,结合常用剂量时人体内所达到的血药浓度,划分细菌对各种抗菌药物敏感、中介或耐药的界限。
14. 野生型(wild-type):最低抑菌浓度不高于流行病学界值的细菌群体,该群体没有获得性耐药及突变耐药。
15. 流行病学界值(epidemiologic cut-offvalue, ECV):区分存在和不存在获得性耐药/突变耐药机制的菌群最低抑菌浓度,通常为野生菌群最低抑菌浓度的上限。
七、参考文献
[1]CFDA. 化学药物非临床药代动力学研究技术指导原则. 2005年3月.
[2]CFDA. 化学药物临床药代动力学研究技术指导原则. 2005年3月.
[3] CFDA. 健康成年志愿者首次临床试验药物最大推荐起始剂量的估算指导原则. 2012年5月.
[4] CFDA.肝功能损害患者的药代动力学研究技术指导原则. 2012年5月.
[5]CFDA.肾功能损害患者的药代动力学研究技术指导原则. 2012年5月.
[6]CFDA.药物非临床药代动力学研究技术指导原则. 2014年5月.
[7]CFDA. 抗菌药物临床试验技术指导原则(第三稿网上征求意见稿). 2014年8月.
[8]CFDA.群体药代动力学研究技术指导原则. 2009年6月翻译.
[9] CFDA. 药物临床试验的一般考虑(征求意见稿). 2015年12月.
[10] CFDA. 药品注册管理办法(修订稿). 2016年7月
[11] CFDA. 药物暴露量-效应关系研究指导原则. 2009年11月译.
[12] CFDA. 药物体内代谢和药物相互作用研究指导原则. 2009年11月翻译.
[13] CFDA. 成人用药数据外推在儿科人群药物临床试验及相关信息使用的技术指导原则(征求意见稿). 2016年11月.
[14]汪复,张婴元.实用抗感染治疗学.人民卫生出版社. 2012年9月第二版.
[15]李耘,郑波,吕媛等. 新抗菌药物临床试验折点制定专家共识. 中国临床药理学杂志,2015,13(11):1067-1074
[16]魏敏吉,赵明,单爱莲. 创新药物临床试验中暴露量-效应关系研究的探讨. 中国新药杂志,2011,20(17): 1608-1611.
[17] 杨进波. 创新性药物临床试验剂量和给药方案的探索和确定. 中国临床药理学与治疗学,2008,13(8): 841-846.
[18] FDA. Guidance for industry: developing antimicrobialdrugs—general considerations for clinical. 1998年7月
[19] EMA. Points to consider on pharmacokinetics andpharmacodynamics in the development of antibacterial medicinal products. 2000年7月
[20] FDA. Exposure-response relationships - study design,data analysis, and regulatory applications. 2003年4月
[21]EMA. Guideline on reporting the results of populationpharmacokinetic analyses. 2008年1月.
[22] EMA. Guideline on the qualification and reporting ofphysiologically based pharmacokinetic (PBPK) modelling and simulation. 2016年7月.
[23] FDA. Guidance for Industry: Safety testing of drugmetabolites. 2008年2月
[24] FDA. Microbiological data for systemic antibacterialdrug products - development, analysis, and presentation (draft). 2009年9月.
[25] EMEA. Guideline on the use of pharmacogeneticmethodologies in the pharmacokinetic evaluation of medicinal products. 2012年8月.
[26]EMEA. Concept Paperon revision of the points to consider on pharmacokinetics and pharmacodynamicsin the development of antibacterial medicinal products 6 (CHMP/EWP/2655/99)and conversion to a CHMP guideline. 2014年2月.
[27] EMEA. Guideline on the use of pharmacokinetics andpharmacodynamics in the development of antimicrobial medicinal products. 2016年7月.
[28] Mouton JW, Dudley MN, Cars O, et al. Standardization ofpharmacokinetic/pharmacodynamic (PK/PD) terminology for anti-infective drugs: anupdate [J]. J Antimicrob Chemother.2005, 55: 601-607.
[29] Gumbo T. Integratingpharmacokinetics, pharmacodynamics and pharmacogenomics to predict outcomes inantibacterial therapy [J]. Curr Opin Drug Discov Devel, 2008, 11(1): 32-42.
[30] Czock D, Markert C, Hartman B, et al. Pharmacokineticsand pharmacodynamics of antimicrobial drugs [J]. Expert Opin Drug MetabToxicol, 2009, 5(5): 475-487.
[31] Gloede J, Scheerans C, Derendorf H, et al. In vitropharmacodynamic models to determine the effect of antibacterial drugs [J]. JAntimicrob Chemother, 2010, 65(2): 186-201.
[32]Martinez MN, Papich MG, Drusano GL. Dosing regimenmatters: the importance of early intervention and rapid attainment of thepharmacokinetic/pharmacodynamic target [J]. Antimicrob Agents Chemother, 2012,56(6): 2795-2805.
[33]Clinical and laboratory standards institute (CLSI). Performance standardsfor antimicrobial susceptibility testing; twenty-first informationalsupplements. M100-S21, 2013, 3: No.1.
[34]Firsov AA, Vostrov SN, Shevchenko AA, et al. Parametersof bacterial killing and regrowth kinetics and antimicrobial effect examined interms of area under the concentration-time curve relationships: action ofciprofloxacin against Escherichia coli in an in vitro dynamic model [J].Antimicrob Agents Chemother, 1997, 41(6): 1281-1287.
[35]Meagher AK, Passarell JA, Cirincione BB, et al.Exposure-response analyses of tigecycline efficacy in patients with complicatedskin and skin-structure infections [J]. Antimicrob Agents Chemother, 2007, 51(6): 1939-1945.
附
抗菌药物PK/PD研究流程