普那霉素IIA 的制备及纯化
Separation and Purification of PristinamycinⅡA
王海燕,李晓露,高月麒,张 丽,任风芝*
(微生物药物国家工程研究中心,河北省工业微生物代谢工程技术研究中心,
华北制药集团新药研究开发有限责任公司,河北石家庄 050015)
摘要:
采用二元液相分离系统,通过对比柱色谱硅胶、聚合物微球和反相硅胶3 种不同类型色谱介质的分离效果和洗脱
参数,开发普那霉素IIA 的制备工艺。结果显示,选用反相硅胶C18 为色谱介质,洗脱剂为乙腈∶ 0.05%磷酸二氢钾溶液(45 ∶ 55),上样量0.8 g/100 ml,纯化所得产品纯度≥ 98.5%,纯化收率≥ 75%。
关键词:
普那霉素;分离纯化;二元液相分离;抗生素;大环内酯;多重耐药菌
普那霉素(pristinamycin) 属链阳性菌素类抗生
素,系始旋链霉菌(Streptomyces pristinaespiralis)产生,由2 种结构和性质不同的物质——普那霉素Ⅰ和Ⅱ组成。其中,普那霉素Ⅰ为环六肽内酯,普那霉素Ⅱ为多不饱和环内酯,两者的组合比例约为普那霉素Ⅱ ( 包括Ⅱ A 和Ⅱ B) 70%和普那霉素Ⅰ( 包括Ⅰ A、Ⅰ B 和Ⅰ C) 30%,其中普那霉素ⅡA( 1) 和Ⅰ A 为主要成分[ 1—2]。普那霉素对革兰阳性菌、部分革兰阴性菌以及厌氧菌均具有很强的杀灭作用,是继万古霉素、替考拉宁之后的又一个抗多重耐药菌的抗生素,而且普那霉素的抗生素后效应较长,耐药性低,不良反应少[3]。
一般采用溶剂萃取、双水相萃取、高速逆流色
谱法等制备普那霉素[4—6],但存在工序复杂繁琐、收率低、不利于溶剂回收等缺陷,而且已有的制备工艺所得产品均为多组分混合物。目前尚未见关于普那霉素单组分的制备方法报道。本研究采用二元液相分离系统,以反相填料为载体,开发了一种操作简单、制备周期短的分离纯化工艺,所得产品纯度高,可为工业化规模生产1 提供参考。
1 仪器与试药
2 方法与结果
2.1 HPLC 法测定1
色谱柱 Agilent XDB-C18 柱(4.6 mm×150 mm,
5 μm);流动相 乙腈∶ 0.01 mol/L 磷酸盐缓冲液(45 ∶ 55);流速 1.0 ml/min;柱温 35 ℃;检测波长 210 nm;进样量 10 μl。
2.2 1 粗品的制备
取1 发酵液100 L,加2 mol/L 盐酸调至pH 3.5,
经板框过滤得到1 滤液。滤液加2 mol/L 氢氧化钠溶液调至pH 5.5 ~ 6.5,再经XAD1600 树脂20 L吸附,用纯化水2 BV 净化树脂柱,以95%乙醇3 BV 解析,将解析液减压浓缩。浓缩液用乙酸乙酯萃取(1 BV×3),合并萃取液,浓缩至0.2 g/ml,冷却结晶,过滤、干燥,得到1 粗品315.5 g。HPLC图谱见图1A。
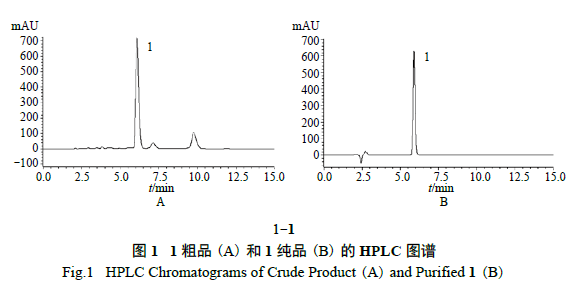
2.3 高压色谱分离纯化
2.3.1 色谱介质筛选
分别选用柱色谱硅胶( 300 ~ 400 目)、聚合
物微球(UniPS 10-300) 和反相硅胶C18(Unisil C18)进行试验。取300 ~ 400 目硅胶100 g,干法装柱,准确称取1 粗品0.8 g,用适量乙酸乙酯溶解、拌样后,加样于硅胶色谱柱顶,用正己烷∶乙酸乙酯( 比例分别为1 ∶ 1、1 ∶ 1.5 和1 ∶ 2)3 BV 洗脱,洗脱流速4 ml/min。分别取聚合物微球和反相硅胶C18 各100 ml 装柱,用乙腈∶ 0.05%磷酸二氢
钾溶液(30 ∶ 70) 平衡。取1 粗品0.8 g,用DMF
4 ml 溶解。分别上样于上述色谱柱,用乙腈∶ 0.05%磷酸二氢钾溶液进行线性梯度洗脱( 0 → 2 h,乙腈30%→ 80% ),洗脱流速6 ml/min。通过色谱检测器在线监测,观察主峰与杂质峰的分离情况,分管收集洗脱液,每管10 ml。采用HPLC 法测定,合并含量( 峰面积归一化法,下同) 大于98.5%的1洗脱液,并按下式计算收率。

每组平行试验3 次,取平均值,结果见表1。
可见,柱色谱硅胶洗脱体积较大,收率低;采用聚合物微球,1 收率有提高,但洗脱体积较反相硅胶相比大得多,且收率也低于反相硅胶。因此,为提高制备效率,减少溶剂用量,选择反相硅胶作为色谱介质。

2.3.2 洗脱溶剂的确定
按“2.3.1”项下确定的介质进行色谱分离,分
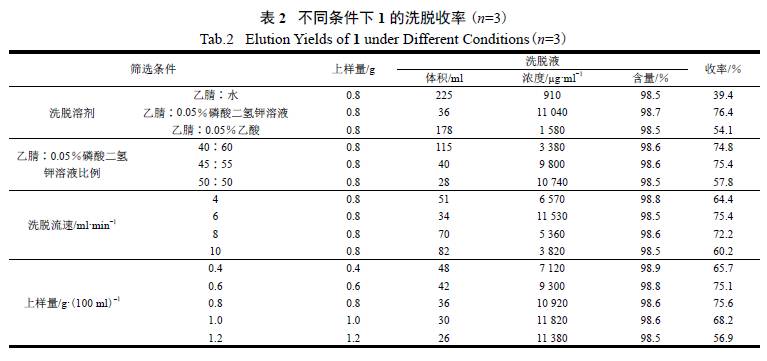
别采用乙腈∶水、乙腈∶ 0.05%磷酸二氢钾溶液、乙腈∶ 0.05%乙酸为洗脱液,按“2.3.1”条件线性梯度洗脱,并计算收率。结果表明,用乙腈∶水、乙腈∶ 0.05%乙酸洗脱时,目标物不易被洗脱,拖尾严重,且主峰与杂质峰不能有效分离,收率低;以乙腈∶ 0.05%磷酸二氢钾溶液为洗脱液时,1 的分离效果较好,杂质去除率、色谱纯度和收率均较高( 表2)。据此选用乙腈∶ 0.05%磷酸二氢钾溶液进行洗脱。
2.3.3 洗脱条件的确定
为提高制备效率和降低能耗,进一步探索等度
洗脱条件。参照“2.3.2”项下线性洗脱结果,当乙腈浓度在40%~ 50%时,主产物被集中洗脱。因此,分别采用乙腈( 浓度40%、45%、50% ) ∶ 0.05%磷酸二氢钾溶液3 BV 等度洗脱,分管收集洗脱液,每管10 ml。采用HPLC 法测定,计算1 含量和收率。结果表明,乙腈浓度为40%时,解吸液拖尾严重,且洗脱率低;当乙腈浓度为50%时,洗脱速度较快,主峰与杂质峰不能有效分离;乙腈浓度为45%时分离效果较好,杂质去除率和1 含量较高( 见表2)。据此确定采用乙腈∶ 0.05%磷酸二氢钾溶液
(45 ∶ 55) 洗脱。
2.3.4 洗脱流速的选择
根据上述优化结果,参照“2.3.3”项下洗脱条件,
分别设定洗脱流速为4、6、8 和10 ml/min,分管收集洗脱液,每管10 ml,采用HPLC 法测定,计算1 含量和收率。结果表明,随着流速的增加,洗脱收率先增后减,当洗脱速度为6.0 ml/min 时,洗脱收率较高( 见表2)。
2.3.5 上样量的确定
根据上述优化结果,参照“2.3.4”项洗脱条件,
分别考察上样量为0.4、0.6、0.8、1.0 和1.2 g/100 ml( 填料) 的分离纯化效果。结果显示,1 上样量低于0.8 g/100 ml 时,溶剂消耗量大,收率低,成本高;随着上样量的增加,收率有所增加;但上样量大于0.8 g/100 ml 时,1 与杂质交叉严重,洗脱收率下降( 见表2)。据此,在保证分离度的前提下,选择上样量为0.8 g/100 ml 填料。










