
为质量控制开发一个恰当的溶出方法需要考虑许多因素,同时要平衡不同的需求。每种药品都有其独特的溶出方法,因为其活性成分、处方、生产工艺都有各自特点。由于人们的认识不同,溶出方法开发与选择的过程仍然具有一定的主观性。
本文为质量控制溶出方法的开发提供了开发策略和具体建议,适用于速释固体口服制剂的质量控制和处方工艺开发。该策略是基于对溶出技术、监管预期、文献和开发各种溶出方法的经验总结。本文也列出了一个采用该策略开发出具有区分力的溶出方法的案例。
关键词:溶出度;口服给药;处方;溶解度;体外模型。
固体口服制剂溶出试验已经使用了几十年以帮助进行处方/工艺开发以及检查并确保药品的批间质量一致性和有效性[1]。
美国药典(USP)通则《溶出试验方法开发与验证》对溶出方法的开发过程进行有价值的说明。USP 通则《溶出度》项下规定了多种溶出试验方法。
有很多文献报道了关于溶出方法开发的研究成果,如 Skoug 等人[2]最早对溶出方法的开发、验证与标准制定进行了综述。Li 等人[3]基于 BCS 分类提出了化合物生物药剂学特性的重要性,并提出了溶出方法开发的决策树。Gray 等人[4]进行了历史性的展望,描述了溶出方法开发和应用方面所面临的挑战。
以上资源为溶出方法开发提供了一个坚实的基础。本文提出了速释固体口服制剂质量控制溶出方法开发的策略,提出了具体的指导和建议,文中所引用的相关文献可作为一些溶出条件和开发考虑的基础。
本案例中的化合物为一个弱碱盐,是采用湿法工艺制备的速释固体口服片剂。溶出的缓冲液由分析级试剂配制。
溶出试验在 Distek 2100C 溶出仪上进行,采用桨法,多种介质,分析采用 LEAP 科技的 OPT_DISS UV 光纤系统,采用 10mm 探针,在 320nm 波长处进行检测。
溶出方法的开发过程如下所示,为便于识别,溶出试验的每个重要参数都被分成单独的段落进行说明。这份开发指南的建立是基于监管部门的建议或指导原则,包括了监管机构所认可的溶出方法的各个方面。
在溶出方法开发中,最重要的是 pH-溶解度数据。溶解度的结果可表明该化合物是否是基于BCS的高溶解性化合物。
如果该化合物的最大规格能在 250ml 的各个介质(EMA指导原则[5]为 pH1-6.8 或 FDA 指导原则[6]为 pH1-7.5)中溶解,那么就被认为是一种高溶解性化合物。
如果该化合物是高溶解性的,应采用 900ml 的 0.1 N HCl、pH 4.5 和 pH 6.8 介质、USP 装置 2(桨法)、50rpm 进行溶出曲线测定。在标准转速下溶出速率最慢的介质可以被选作溶出方法。较慢的溶出速率将能够增加区分处方组成、生产工艺变化或药动学性能的可能性。适当的溶出介质可以确保该方法适合测定一个 BCS 3 类或 BCS 1 类的高溶解性化合物在整个货架期间内是否符合标准规定。
如果该化合物在上述 pH 范围内溶解度低,那么最初目标是开发一个在 30~60 min 至少达到 85% 溶出量的溶出方法[7]。如果采用不同粒度的 API、不同剂型或其他关键属性进行了动物 PK 研究,并发现了 PK 的差异,那么该方法理应能够对这些处方进行等级划分。本文后续部分对如何为低溶解性化合物开发一个适当的溶出方法提供了指导。
对于速释固体口服制剂制剂,通常采用 USP 装置 1(篮法)或装置 2(桨法),其他 USP 装置通常用于控释或非口服制剂。
除非特殊情况,如果预期的商业剂型是非漂浮剂型,那么应该选择桨法。
对于崩解型处方,篮法潜在的问题是其转篮下方的流体动力学环境不像桨法那样能够充分混合,这将会导致溶出数据的分析更加具有挑战性。
有时在药物的早期研究阶段使用的剂型可能与最终商业剂型不同,如果早期的剂型(如胶囊)漂浮,那么应考虑使用沉降装置,以便于以后使用桨法。这将为胶囊转化为片剂后,在方法中去掉沉降装置的溶出数据提供一种最简单的数据追踪方式。
在所有阶段,这都是一个很好的能减少溶出方法变更的策略。对于一个漂浮剂型,应评估篮法和带沉降装置的桨法。有时,篮法在非崩解剂型上有一定优势,篮法能够提供相同的流体动力学环境同时也能够保证介质充分接触到该制剂。
根据 FDA[7]、EMA[5] 和日本医药食品安全局(PFSB)[8]规定,桨法,50 rpm 应作为溶出方法开发的起点。
如有圆锥形堆积(不溶解性辅料堆积在桨下,限制介质进入内部),应考察 75rpm。FDA 和 PFSB 推荐 75rpm 作为选项。增加桨的转速可使辅料分散的更好,模拟体内分散,使溶出不受阻碍。如果要使用 100rpm 桨法,应有充分的理由,例如可以避免使用表面活性剂或减少表面活性剂的使用浓度。
采用篮法时,应首先考察 100rpm 的转速[5,7,8]。FDA[7] 也增加了篮法 50rpm 选项,如果溶出太快不能提供一条有区分力的曲线,可以考虑 50rpm 篮法。
只有当指导原则推荐的转速参数不适用的情况下,才可考虑使用其它转速,使用其它转速应有充分证据。选择一个不同的介质(例如,对这个化合物具有较高溶解度的)来加快溶出比增加转速更好。
在溶出方法开发中一个好的诊断工具是在溶出试验结束后增加“无限转速”,以使颗粒被强制分散同时使未溶解的API的溶解。
例如,可在最后一次正常取样点后,将转速增加至 150~250 rpm 维持 15~30 min,再进行取样。
这种做法可以快速检查制剂的含量,并确保溶出不是受到溶解度的限制或者低的溶出量不是由含量低引起的。然而对于批放行检验,转速的增加值是受到限制的。
在处方开发过程中,使用生物相关介质进行溶出试验对内部决策具有重要作用,而常规的 QC 试验则可以采用一种完全不同的方法[9]。生物相关介质用于模拟复杂的人体胃肠道溶液,常常用于方法开发过程中,以对一个化合物体内的溶解性和稳定性进行更多的了解,同时也用于处方筛选。
Johann Wolfgang Goethe 大学的 Jennifer Dressan研究了人体胃肠道溶液的复杂性,发表了大量关于生物相关介质及其评价的文章[10-15]。
需要注意的是,采用生物相关介质的溶出方法并不具备生物可预测性(与化合物临床行为相关),除非在临床研究数据中已经建立了这种关系。如果 QC 试验采用生物相关介质,那么将会导致成本高、资源需求大和复杂性(发生错误的可能性)等问题。采用生物相关介质会增加耐用的质量控制溶出方法开发的难度。
因此,简单的缓冲盐体系对于常规溶出分析而言更受欢迎,根据化合物自身特性所建立的方法可能具备生物可预测性。
溶出介质种类和体积需要满足漏槽条件。
USP 将漏槽条件定义为:一种药物,其溶出介质体积至少是能形成其饱和溶液体积的 3 倍。pH-溶解度曲线对于初步筛选溶出介质提供了最有效的信息。Skoug 等人[2]提出,体内体外相关性可能在接近饱和溶解度时达到。
然而,当溶出方法接近这个溶解度限度时,该试验过度敏感的风险也随之上升,同时方法的耐用性也随之下降。开始时采用相对较慢的溶出速率(仍然要求在 60min 不低于 85% 的溶出量)筛选溶出方法不但可以提供足够的区分力,而且可以降低后期开发阶段必须采用更加灵敏的试验来评价处方或者工艺变更的风险。
溶出介质的 pH 范围一般为 1.1~6.8。由于溶解度原因,需要更高pH的溶出介质时也可以接受,但通常不得超过 8.0 [7]。
溶出介质基于所需的 pH 来选择:
溶液 pH 为 1.0~3.0 选择盐酸
pH 2.0~3.0 选择甘氨酸
pH 2.5~3.5 选择柠檬酸盐
pH 4.0~5.5 选择醋酸盐
pH 6.0~8.0 选择磷酸盐。
一个典型的缓冲盐溶出介质应具备 0.05 摩尔浓度。纯水由于其来源不同,pH 会不同,一般不作为溶出介质。
如果在上述 pH 范围的介质不能提供足够的溶出量,那么应该评估是否使用表面活性剂。十二烷基硫酸钠(SDS)也叫做十二烷基磺酸钠(SLS),是首选的表面活性剂[7]。SDS是介质配制最常用的表面活性剂,是因为其纯度高,不同厂家间质量基本一致,而且很容易准确配制至所需的浓度。SDS在低于 pH2 的条件下会降解,但仍然可成功地在这些介质中发挥作用[16]。
由于上述原因,SDS 需要有一定稳定性或具备一定的使用期限。SDS纯度应大于 98%,目前可接受的SDS等级和其它监管中使用到的表面活性剂可以在 FDA 溶出网站上查到[16]。
溶出试验中使用的表面活性剂浓度应进行合理说明,一般选择能达到溶出曲线接受标准的最低浓度。0.1%~3% 或者更高的 SDS 浓度是可以接受的[16]。0.23% SDS 水溶液像一个润湿剂,而不是一个溶解溶剂,因为该浓度低于其临界胶束浓度。如果在如前所述的可接受的 SDS浓度下,仍然无法在 60min 溶出 85% 以上,或者与活性成分或辅料发生反应,那么应该选用 FDA 网站[16]上的其他表面活性剂。
溶出介质的脱气效果应该进行研究。室温下的溶出介质会比 37℃ 时溶解更多的气体。当对溶出介质进行加热时,这些溶解的气体会形成气泡,可能会对溶出产生不可预测的影响。气泡可能导致制剂粘附在溶出杯上,能够减少介质与颗粒接触,或者增加颗粒漂浮的倾向。这些都会导致溶出速率发生显著变化。
制药工业和监管部门可接受的标准溶出介质体积一般为 500ml 和 900ml。选择这个体积是为了使化合物达到漏槽条件,而不是代表药品在体内所接触的液体体积。McConnell 和 Mudie 发表过关于人体胃肠道生理特性的详细综述[17,18]。
在开发阶段可以使用较少的溶出介质体积以减小对 API 的需求。如果有漏槽-溶解度方面的原因,那么可以用更大的体积,如 4L。无论哪种情况,一个合适的装置都是必须的。制剂自身的溶出特性(如变异性和溶出曲线)是选择介质体积的最佳指导。尽量在以上这两种标准介质体积中选择,这样有利于方法转移也有利于减少监管方面出现问题。
一个药品的所有规格最好使用同一种方法和相同的介质体积,这样可以对各个规格的溶出曲线进行评估,也能对相同或相似处方进行溶出评价。这种方法可以揭示高规格和低规格是否在体外溶出相似。此外,不同规格使用相同的溶出方法,可以为在特定情况下只研究高规格和低规格提供便利。
在速释制剂溶出方法开发时,取样时间点一般从 5min 到 60min 以上。5min 点可用于混悬剂或其它快速溶出剂型,此时变异不高。典型的取样时间为 15、30、45、60min。
然而,取样时间点取决于产品特性和方法追踪处方核心属性的能力。如果希望更好的研究或追踪崩解效应,5min 或 10min 时间点都是需要的。对于快速溶出产品,光纤溶出系统可以通过更多的取样时间点得到更好的溶出曲线。如果在减慢溶出液稳定性方面有显著的风险,那么应在 60min 后增加一个取样时间点以保证至少获得 85% 的平均溶出量。
一般来讲,15min 点是很有用的,特别是化合物在这段时间内已经溶出至少 85%。
如果是在 0.1N HCl 中满足这种情况,FDA[7] 认为这个制剂的行为与溶液一样,通常不应该有任何生物利用度方面的问题。15min这个时间点也是 BCS 3 类药物可能 BE 豁免的一个指定的时间点[5]。
最后,对于在15min 内溶出至少 85%[6] 或大于 85%[5] 的溶出曲线,相似性评价时不需要采用 f2 因子法。
如前所述,在 USP 装置 2(桨法)测定胶囊时可以使用沉降装置。此外,对于有粘性的片剂和崩解缓慢的片剂也可以使用沉降装置。
当片剂粘在溶出杯的非中心位置上时,溶出曲线可能会产生高变异结果。不在中心的片剂与位于中心的片剂相比处于一个不同的流体动力学环境中[19,20]。
缓慢崩解的片剂需要周围液体流动来产生更加一致的溶出曲线。使用桨法进行测定时,将这种片剂装在沉降装置里可以解决这些问题。应该研究各种沉降装置类型确定哪种适合,在方法中应该规定使用的沉降装置的类型,因为不同类型的沉降装置在制剂周围具有不同的流体动力学环境。
对溶出样品进行过滤不但可以减少取样后 API 颗粒的溶出,还可以降低辅料颗粒堵塞分析设备的风险。典型的滤膜孔径是 0.45~70 µm。对于微粉化药物而言,分析者应尽可能使用最小孔径的滤膜。
溶出分析方法取决于药物的剂型、药物量、药物的 UV 吸收情况。采用光纤分析或在线 UV 对溶出样品进行检测是有效的技术手段,在溶出试验结束时就能提供结果,加快了速度。UV 方法是否适用,取决于合适的吸光度和没有明显胶囊壳或辅料干扰。HPLC/UPLC 是另一种典型的分析方法,在 HPLC 分析中,对低规格药物最好增加进样体积而不是通过减少溶出介质来增加检测灵敏度。
溶出参数(如 pH、表面活性剂浓度等)变化对溶出曲线的影响应该在方法的开发阶段进行评估。
例如,在溶出耐用性考察中,pH 值的微小变化影响应该进行评估,±0.05 的偏差不应该对溶出速率产生显著影响。如果有显著影响,那么应该根据耐用性结果选择其他合适的 pH 值。
通常采用变更目标商业规模处方或工艺参数所制备的批次来评价溶出方法的灵敏度或区分力。需要考虑的因素包括:药物粒径、处方组成(如低量崩解剂)、工艺变化(如过度制粒)。这些变化因素可以采用拟定溶出方法进行评价,应该证明在体外具有足够的区分力。然而,什么才是足够敏感是因项目而异的。
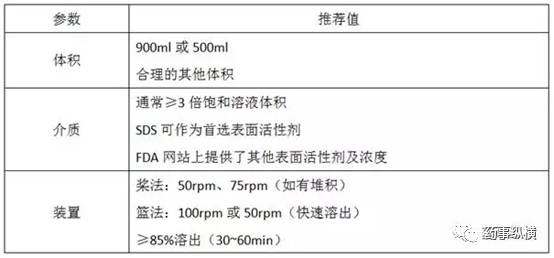
表 1 汇总了在溶出方法早期开发阶段的一些关键建议。溶出方法开发应基于对关键处方/工艺参数变化的灵敏度以及不同处方的临床PK研究结果进行持续优化。在开发阶段,处方调整或其他处方可能在临床中进行测试。
在研究体内体外相关性(IVIVC)的溶出方法优化中,经常需要临床PK结果。要通过调整溶出结果使其更好的重现体内结果来优化溶出试验。
表 1. 溶出试验参数和推荐值
需要注意的是,当在研究中采用两个相似处方时,不应期望溶出试验一定能够模拟体内趋势。这是因为每一个处方的关键处方变量可能不同,使用一个通用的 IVIVC 来用于这两个处方评价可能不能实现。如果一个特定处方的调整在临床研究中进行了测试,这些结果可以用来指导调整溶出方法来建立体内体外相关性(IVIVC)。IVIVC的开发策略、IVIVC 获批后如何测定、以及批选择的过程在最近的一篇文章中进行了详细描述[21]。
下面列举了一个经湿法制粒的速释固体口服片剂的溶出方法开发案例。
这个化合物是一个弱碱盐,pH-溶解度曲线见图 1,这个化合物仅在酸性条件下溶解。对于一个标准的速释固体口服制剂来讲,API 的粒度对处方是一个关键质量属性。通过两个不同粒径的API的片剂的研究,进行了介质的选择,见图 2。
在不同 pH 的介质中,采用 50rpm、900ml 进行了研究。结果表明, pH3.0 是一个符合漏槽条件、对 API 粒径具有区分力的溶出介质。
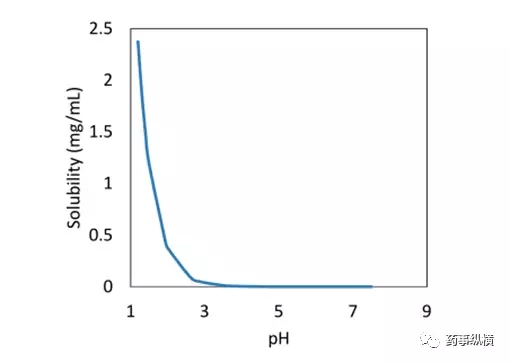
图1. 化合物盐形式的 pH-溶解度曲线
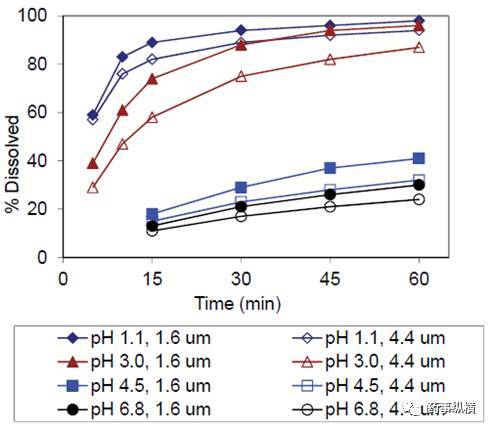
图 2. 不同粒度 API(D50=1.6和4.4µm)的片剂在不同 pH 介质下的溶出曲线(900ml,50rpm)
第二个需要研究的关键质量属性是这个化合物的物理状态。这个化合物是盐的形式,遇湿会可能会转化成游离碱形式。通过不同比例游离碱盐制备的两个批次片剂对不同 pH 介质的区分力进行了研究,见图 3。
pH 3.0 是最具区分力的介质,同时也满足在 60min 至少溶出 85% 的开发标准。pH 3.5 介质在这个时间内溶出没有达到至少 85%。pH 3.0 介质溶出谱能够区分出很宽比例的游离碱盐的片剂,见图 4。
随着比例的增加,溶出速率下降。采用 50rpm,900ml,pH3.0 介质这个溶出条件对不同批次的考察结果表明,对处方中这两个主要关键属性是具有灵敏度或区分力的。
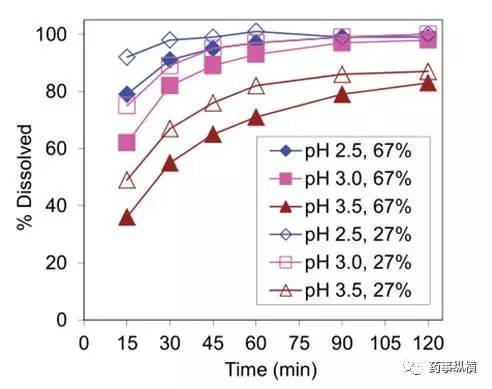
图3. 两种不同API比例27%和67%的片剂在不同pH介质中的溶出曲线(900ml,50rpm)
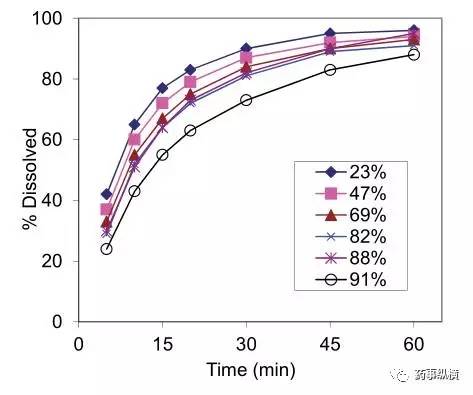
图4. 不同API比例的片剂的溶出曲线(溶出方法:50rpm,900ml,pH3.0介质)
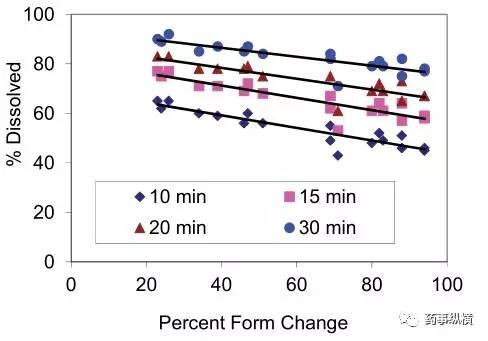
图5. 不同API比例和溶出量的关系(溶出方法:50rpm,900ml,pH3.0介质)
本文对质量控制溶出方法的开发提出了一项策略和具体建议。溶出方法的开发基于化合物及其处方的特性。这项开发策略是基于世界各地健康监管部门的法规。
一个速释固体口服制剂的溶出方法开发案例表明,对关键质量属性应具有一定的区分力。一个溶出方法的优化应基于其对关键处方/工艺变化的敏感性,如有可能,应尽可能反映出真实的体内响应。
原文名称:A Strategy for Quality Control Dissolution Method Development for Immediate-Release Solid Oral Dosage Forms 点击原文链接可下载英文原文。

声明:本文为药事纵横小编编译,请尊重小编的劳动成果,转载本文务必获得药事纵横许可,否则一律视为恶意侵权。
药事纵横——药品研发的信息窗,我们的网址是www.pharmaguider.cn, 点击原文链接可直达我们的首页,我们期待您的光临。

药事纵横是一个开放,由自愿者组成的团体,现有成员14名,分别为Voyager88,雷诺岛,三分话,Herman,Mzwinsunny,文竹,duke,巉巉之石,蓝色枫叶,ISAL,海角边,冯姝婷,yhqqqqq,陈小牛,欢迎有志之士加入我们团队。投稿、加专业微信群【合成、制剂、分析、注册、BD、一致性评价】请加微信442015666,QQ群:22711855/22711679(限加一个)










