
摘要
:
干细胞研究和临床试验发展迅速,目前全球干细胞临床研究排名前三的国家或地区分别是美国、欧洲和中国,干细胞治疗种类以造血干细胞为主,间充质干细胞日益增长,神经干细胞和多能干细胞的临床试验也相对较多。目前全球已有
14
款干细胞药物上市,超过一半以上是间充质干细胞治疗产品。中国共有
87
个干细胞临床项目完成备案,其中间充质干细胞备案项目最多。细胞治疗是按照医疗技术还是药品来监管,世界各国有所不同。在美国按细胞组织类产品风险高低进行归类监管,欧盟以先进技术治疗医学产品归类监管,日本按照再生医学产品管理,中国目前进行机构和项目双备案制度。在严格分类管理的基础上,无论欧盟的医院豁免制度、日本的条件限制性准入政策,还是中国从药品
-
第三类医疗技术
-
备案制管理政策的变迁,都为干细胞及其他细胞治疗产品的研究和应用提供了科学而快速发展的政策保障。对国内外干细胞临床研究及应用的发展现状进行综述,同时分析各国干细胞临床研究相关的法律法规与质量控制监管政策。
干细胞是一类具有自我更新和多向分化潜能的细胞,在个体发育和疾病发生中扮演重要角色,也是再生治疗中的关键
“
种子细胞
”
。干细胞具有再生、替代、修复和分化能力,在生命科学及医学领域的应用前景广阔。
本文对国内外干细胞临床研究及应用的发展现状进行综述,对干细胞临床应用的前景进行展望。同时,分析各国干细胞临床研究相关的法律法规与质量控制监管政策,以期对中国干细胞临床研究及应用规范有序地开展提供有益借鉴。
1
全球干细胞临床研究及应用的发展现状和趋势
干细胞治疗一直是生命科学前沿最受重视的领域之一,目前全球干细胞研究和临床试验正在如火如荼地开展,每年有大量新增临床研究项目,整体呈上升趋势(图
1
)(数据来源
https//www.clinicaltrials.gov
)。
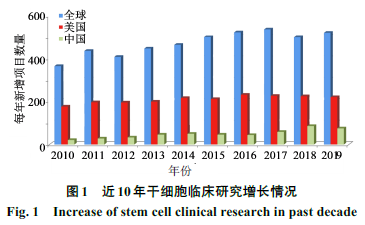
截止到
2020
年
7
月
31
日,用
stem cell
作为关键词,在
ClinicalTrials.gov
网站检索到
8 049
项登记注册的干细胞临床试验方案。按国家和地区统计,目前(截止到
2020
年
7
月
31
日)全球干细胞临床研究排名前三的国家或地区分别是美国(
3 986
项)、欧洲(
1 902
项)和中国(
645
项)。新的研究领域和新方法的推动,以及各国政府政策的支持,促进了干细胞治疗的快速发展。
从疾病治疗领域来看,神经系统疾病、癌症和肿瘤类疾病、出生前疾病和异常、血液和淋巴系统疾病、心血管疾病是目前临床研究数量较多的疾病领域。在干细胞治疗的细胞种类选择上,造血干细胞(
hematopoietic stem cell
,
HSCs
)的临床试验数量最多,占比近总数的
50%
(
3 898/8 049
),体现出
HSCs
在干细胞临床研究中受到的高度关注;间充质干细胞移植临床试验共
1 155
项,在近几年数量持续增长,说明间充质干细胞的重要性日益增强
[
1
]
。在其他类型细胞中,神经干细胞和多能干细胞的治疗研究进入临床试验阶段的项目数量也相对较多,其中神经干细胞被主要应用于中枢神经系统疾病的治疗,而多能干细胞被主要应用于眼部疾病和遗传性疾病的治疗。
1.1
目前全球已上市干细胞药物
全球干细胞临床试验进展情况显示,目前
45%
干细胞临床试验处于早期阶段,
II
期临床试验比例为
22.5%
,验证性
III
期临床试验仅占
4.9%
。目前全球已有
14
款干细胞药物上市(表
1
)(数据来源
https//www.clinicaltrials.gov
),适应证包括膝关节软骨缺损、移植物抗宿主病、克罗恩病、骨修复、急性心肌梗死、遗传性或获得性造血系统疾病、退行性关节炎和膝关节软骨损伤、克罗恩病并发肛瘘、赫尔勒综合征、肌萎缩性侧索硬化症、中度至重度角膜缘干细胞缺乏症、血栓闭塞性动脉炎等疾病。在获批上市的干细胞药物中,超过一半以上是间充质干细胞治疗产品。根据
Polaris Market Research
发布的最新研究报告,
2018—2026
年全球间充质干细胞市场预计以
7.3%
的复合年增长率增长。同样,全球市场调研机构
ARC
(
AnalyticalResearch Cognizance
)发布的报告也显示,全球间充质干细胞市场发展迅速,预计至
2024
年底其市场值将达到
2.2
亿美元
[
2
]
。

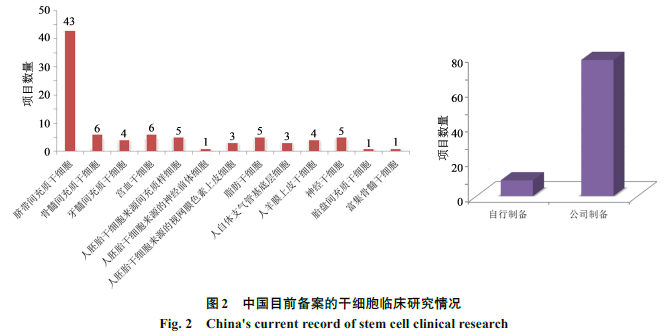
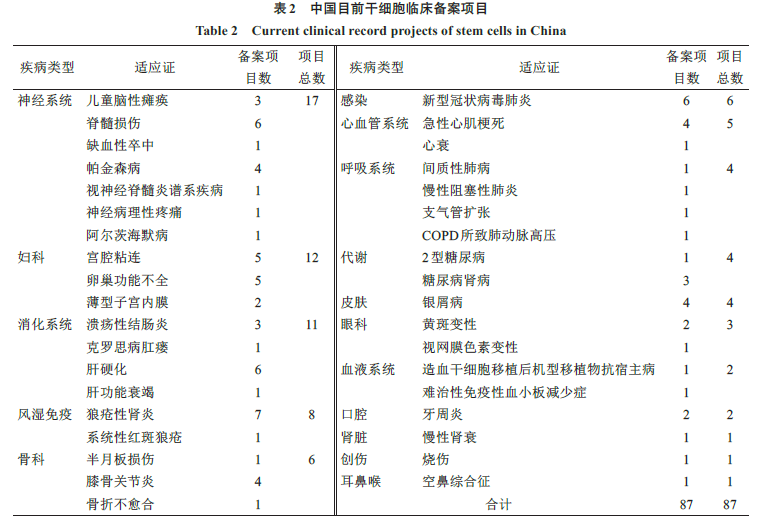
中国干细胞研究水平已经跻身世界前列。到目前为止,中国共有
108
家干细胞临床备案机构,覆盖
26
个省(市);共有
87
个干细胞临床项目完成备案,涉及
55
家机构,覆盖
21
个省。中国临床研究备案的细胞种类包括间充质干细胞、胚胎干细胞、神经干细胞等,其中间充质干细胞备案项目最多,涉及脐带、骨髓、胎盘、牙髓、脂肪等来源的间充质干细胞,致力于神经系统、妇科、消化系统、风湿免疫病、骨科、感染、心血管系统等多种疾病,临床应用前景广阔(图
2
、表
2
)。
2
干细胞临床研究及应用的管理政策
世界各国对于干细胞临床研究及应用的监管经历了数十年变革,处于不断完善修订中。干细胞作为体细胞治疗的一种产品,在遵循体细胞治疗的管理规定上有所补充。大多数国家的干细胞治疗由药品管理部门管理,少数国家实行双轨制。
2.1
美国
1993
年美国食品药品监督管理局(
FDA
)出台了《人类体细胞治疗和基因治疗指导原则》,但并没有统一的管理标准,建议逐案处理(
case by case
)。
1997
年美国
FDA
向联邦政府提交了《对人体细胞及组织产品的管理建议》,建议将细胞治疗纳入美国药品法规,接受美国
FDA
监管。
2005
年《人体细胞及组织产品的管理规定》作为联邦政府的正式法规颁布并收录在联邦法规的第
21
章
1271
条,简称《
21 CFR 1271
》,一直沿用为美国目前对干细胞治疗管理的主要法律依据,其依据细胞组织类产品的风险高低进行分类管理:低风险类产品(符合《公共卫生法》
361
条规定)不需上市前风险评估,可在医院直接进行临床应用,机构接受定期检查,但检查前即可对外提供产品;高风险类细胞组织类产品(符合《公共卫生法》
351
条规定)按生物制品或新药或医疗器械管理,需要上市前经
FDA
评估和许可,申请获批前后均要对机构进行检查。《
21 CFR 1271
》规定美国细胞治疗遵守的规范主要包括《人体细胞组织现行优良操作规范》(
Current Good Tissue Practice
,
cGTP
)和《药品现行良好生产规范》(
Current Good Manufacture Practice
,
cGMP
)等,遵循
cGTP
、
cGMP
及临床试验规范(
GCP
)指南是监管的关键,尤其对于活化,扩增或基因修饰的细胞等高危治疗,确保细胞产品的安全性和有效性。
2.2
欧盟
在
1993
年《医疗器械法》和
2001
年《医药产品法》的基础上,欧盟药品管理局(
EMA
)于
2007
年颁布并于
2008
年
12
月
30
日正式实施的《先进治疗医药产品管理规定》成为干细胞治疗管理的法律依据,将基因治疗医药产品、体细胞治疗医药产品和组织工程产品纳入先进治疗医药产品
ATMPs
(
AdvancedTherapy Medicinal Products
)管理,要求
ATMPs
在欧盟各国上市需遵循集中审批程序,由欧盟
EMA
负责审批,要求设立先进的治疗委员会,建立相应的
GMP
和
GCP
标准。同时该法规中提出了医院豁免条款,由医院决定对患者的治疗应用。考虑到
ATMPs
上市后的风险,特别强调此类产品的可追踪性,要求生产商和医院均需建立相应的追踪制度,生产商要保证生产、包装、贮存、运输和配送以及原料来源的可追溯性,医院要保证产品和患者的可追溯性。欧盟
EMA 2004
年颁布的《人类组织和细胞捐赠、获得、筛查、处理、保存、贮藏和配送的安全和质量标准》,对人体组织和细胞应用程序的每一个步骤,都提出了安全和质量技术要求,成为
ATMPs
产品的主要技术指南。
2017
年
11
月
28
日《先进治疗医药产品生产质量管理规范指南》成为欧洲细胞治疗遵守的规范。
2.3
日本
日本
2013
年修订了《药事法》,将其更名为《药物、医疗器械与其他产品法》,以此为依据,将细胞治疗、基因治疗和组织工程产品作为独立于药物和医疗器械的再生医学产品进行单独监管。
2013—2014
年相继出台了《再生医学促进法》和《再生医学安全法》,为研发和临床应用提供了法律依据。《再生医学安全法》允许企业将产品注册为低、中、高
3
类风险之一。再生医学产品实行双轨管理,在原有药品审批基础上,允许条件性准入某些经过临床试验安全有效的治疗方法,可更快实现商业化审批,最长条件准入时间为
7
年,期间收集有关疗效数据,到期后再次进行申请长期上市或者退出市场。厚生劳动省颁布了一系列指南对细胞治疗进行规范,包括《干细胞临床研究指南》《人体自体细胞
/
组织产品质量控制与安全指南》《细胞组织操作原则》等
[
3
]
。
2.4
中国
为了规范干细胞临床研究及应用,促进干细胞治疗技术科学有序地发展,中国陆续出台了一系列政策和法规。
1993
年
5
月,原卫生部发布《人的体细胞治疗及基因治疗临床研究质控要点》
[
4
]
,首次将人的体细胞治疗纳入药品管理;
1999
和
2003
年先后颁布《新生物制品审批办法(局令第
3
号,
1999
)》和《人体细胞治疗研究和制剂质量控制技术指导原则》
[
5
]
,明确人体细胞治疗按新药注册,要求每个方案的整个操作过程和最终制品必须制定并严格执行标准操作程序,以确保体细胞治疗的安全、有效。
2009
年
3
月,原卫生部发布《医疗技术临床应用管理办法》(卫医政发〔
2009
〕
18
号)
[
6
]
,将细胞治疗划分为第三类医疗技术,对其实施准入管理。规定第三类医疗技术由卫生部负责技术审定和临床应用管理,研究机构证实动物试验和临床试验有效,提交申请给卫生部,经卫生部审定批准后再用于临床治疗,中国的体细胞治疗全面放开。
2010
年,原卫生部审议通过了《药品生产质量管理规范》(
2010
年修订)》,贯彻质量风险管理和药品生产全过程管理的理念,与世界卫生组织的
GMP
规范接轨,成为细胞在实验室制备过程中必须遵循的规范。
2015
年
7
月,国家卫生计生委下发《国家卫生计生委关于取消第三类医疗技术临床应用准入审批有关工作的通知》,正式取消第三类医疗技术临床应用准入审批。
2016
年
10
月国家卫生计生委与国家食品药品监督管理总局(
CFDA
)[现国家药品监督管理局(
NMPA
)]共同组织制定了《干细胞临床研究管理办法(试行)》
[
7
]
和《干细胞制剂质量控制及临床前研究指导原则(试行)》
[
8
]
,中国施行干细胞临床研究备案管理制度,干细胞治疗从人的体细胞治疗中分类出来,干细胞临床研究有了专门的实施依据。
2017
年
3
月,国家卫生计生委和
CFDA
(现
NMPA
)在其医学研究备案登记信息系统中公布了首批通过备案的
8
个干细胞临床研究项目。同年
10
月,
CFDA
(现
NMPA
)发布《生物制品注册分类及申报资料要求(试行)》,要求将细胞治疗技术按照治疗性生物制品进行申报。同年
12
月,发布《细胞治疗产品研究与评价技术指导原则(试行)》
[
9
]









