背景:
靶向核酸酶改变了基因组编辑技术,为哺乳动物基因组定向编辑提供了有效的方法。与此同时,对Cre-LoxP技术的需求不断增加,Cre-LoxP作用于复杂的基因操作如:多基因删除、增加,基因融合和一定条件下特定位点基因序列的移动。
结果:
我们分别使用两套高效病毒系统,整合慢病毒和非整合腺病毒结合病毒,设计并构建了两套Cre重组酶表达载体。我们揭示了Cre转入携带LoxP红色荧光蛋白和嘌呤霉素抗性报告基因的稳定重组HEK293细胞系的效率。转入的Cre重组酶能够有效地直接或在一定条件下剪切LoxP红色荧光蛋白和嘌呤霉素基因,由此证明了这些分子工具的功能。由于两个筛选标志物的方便使用,这些基于病毒的体系为更先进的基因编辑、复杂的基因组杂交到各类型的细胞和不同情形提供了一种稳健且方便使用的工具。
结论:
我们开发且有效地验证了两种基于病毒的Cre重组酶转移体系,可有效用于多种哺乳动物细胞的基因组操作。内置报告基因和可诱导的基因元件使得基因转移很容易,能够用于活细胞监测、药物筛选、暂时的基因敲除、扩大基因编辑的使用范围。
有效的靶向核酸酶,如:归巢核酸内切酶、锌指核酸酶(ZFN)、类转录活化因子核酸酶(TALEN)和常间回文重复序列丛集/常间回文重复序列丛集关联蛋白系统,为基因组工程提供了现代的工具。显然,靶向的核酸酶不仅可以在一个复杂的哺乳动物基因组中和特定的基因位点结合,而且可以通过它们的核酸酶活性形成双等位基因的改变。这个新的特性可以用于有效地构建基因敲出动物且能直接用于多个物种如人类细胞和疾病细胞系。事实上,和同源重组联用,靶向核酸酶已经被用于基因组编辑,通过使用构建的细胞模型,阐明了许多生物学和疾病进展。然而,单独使用靶向核酸酶只能产生双链DNA断裂,这又反过来在结合位点附近刺激易出错的非同源末端连接(NHEJ),造成小片段的序列改变,也叫变异。和Cre-LoxP技术联用,靶向核酸酶可以潜在地用于更先进、复杂的基因组编辑形成大片段且准确的改变如框架融合或者单核苷酸修正。
Cre-LoxP是一个位点特异的重组酶技术,被用于实现转基因小鼠特定位点的基因删除、插入和倒位。Cre-LoxP包括一个核酸酶,Cre重组酶来源于P1噬菌体,可以识别两个LoxP之间的识别位点并催化重组。每个LoxP位点包括34bp的共有序列,包括一个8bp的核心间隔区域附于两个13bp的反向重复序列的任一边。当一个DNA元件被两个LoxP位点侧面附着后,Cre重组酶可以识别LoxP位点并剪切。两个位点间的DNA元件被移除,附着的位点结合在一起。LoxP位点的朝向和位置决定了Cre重组酶诱导删除、倒向或染色体移位。这些可预测的变化可用于形成一定条件下的敲出以及报告基因的融合并用于研究基因的功能和调控。传统地,Cre-LoxP体系是制作条件敲出小鼠的主要方法。通过同源重组,LoxP序列首先被加载至目标基因的侧面。通过表达Cre重组酶可以随后筛选出条件性基因敲出纯合子后代。虽然Cre-LoxP体系一直被用于转基因动物,但是最近几年它才和靶向核酸酶一起被用于体外培养的哺乳动物细胞。这是因为根据不同应用,Cre和靶向核酸酶(ZFN,TALEN,Cas9)各有优缺点。Cre仅能编辑侧面含有LoxP的序列,但是它在移除或倒向侧面含有LoxP的序列的可预测性、精确性和稳健性不能被缺乏特定性和精确度的靶向核酸酶替代。因此,当侧面含有LoxP的序列存在时,增加Cre可以增加基因组编辑的可能性并能实现更复杂更先进的基因组操作。然而,缺少一个稳健易用的Cre传递体系使得它的实际应用受阻。
为了解决这个问题,我们用两种非常有效的病毒基因传递方法慢病毒和腺病毒结合病毒技术(AAV),与Cre-LoxP基因编辑技术结合。经证实,这两种病毒体系都能在有显著特征的多种哺乳动物细胞类型中传递转基因。慢病毒的整合性使得基因传递非常高效,而AAV则是潜在的治疗应用中更安全的方法。为了使用两种体系,我们设计并构建了两套Cre重组酶表达载体。为了能够实现活细胞监测和改造后细胞的富集,我们更进一步设计了携带绿色荧光蛋白或嘌呤霉素的两套载体。
下面,我们报告两种新的基于病毒的Cre传递体系在体外培养哺乳细胞中的进展和潜在的应用。使用之前构建的TALEN编辑的人HEK294细胞,我们证实了两种Cre传递体系能够移除侧面含有LoxP基因的序列。我们的研究发现在HEK293细胞里由病毒转导的Cre重组酶可以成功地移除由靶向核酸酶和同源供体引入的侧面含有LoxP基因序列。这一新的资源可以为科研工作者实施更复杂更先进的基因组工程提供一种强健且易使用的方法。
为了给基因编辑构建一种强健且易使用的Cre转导体系,我们在这项研究中用了三种顶尖的技术:i)靶向核酸酶(TALEN)对双链DNA的精准剪切能力ii)Cre-LoxP位点特异的重组酶iii)两种有效的病毒基因转导体系,慢病毒和AAV转导。
首先,我们设计并构建了五个可以表达一个或两个启动子的Cre重组酶的慢病毒载体(图1 a-e)。一个载体包含一个驱动Cre表达的CMV启动子和3’末端的WPRE (图 1a)。三个载体都是双启动子,一个CMV启动子驱动Cre表达,一个EF1α启动子和WPRE在标记基因的3’末端驱动GFP和/或嘌呤霉素表达(图1 b-d)。第五个构建是一个可诱导的载体,Cre被克隆到一体化的Cumate开关型慢病毒载体(图 1e)。在这个Cumate可诱导的载体中,Cre由Cumate开关(CuO)驱动,一旦添加Cumate后开关被打开(System Bioscience Inc.(SBI),Palo Alto,CA),EF1α驱动CymR抑制子和嘌呤霉素的表达,CymR抑制子和嘌呤霉素由一个核糖体跳过点T2A分隔开(图 1e)。对于慢病毒载体,WPRE mRNA稳定性序列位于poly-A信号的5’端。
用类似的方法,表达Cre的AAV载体从5’到3’端依次为:5’末端反向重复序列,一个组成性的CMV,Cre,一个poly-A信号,和3’末端反向重复序列(图1 f-i)。活细胞监测和/或药物筛选的标记基因GFP和/或嘌呤霉素被包含在组成性的EF1α启动子中(图1 f-i)。在包含两个标记基因的构建载体里,T2A位于GFP和嘌呤霉素之间。
在功能实验检测之前,以上表达载体的真实性和准确性先由全序列测序检测。
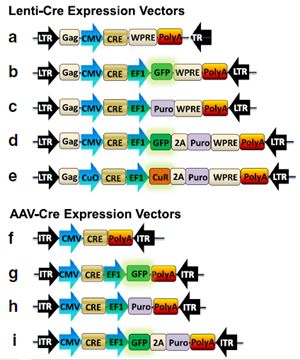
图1.表达Cre的重组AAV和慢病毒载体设计和构建图示说明。慢病毒载体的特征描述了人源化的在没有选择标记的CMV启动子控制下的Cre基因(a)或者与可选择的筛选标记如GFP结合(b)嘌呤霉素(c)GFP和嘌呤霉素(d)一体化的慢病毒载体里Cumate可诱导的CRE表达(e)类似地,AAV载体的特征描述了单独在CMV启动子控制下的人源化的Cre基因(f)携带可选择的筛选标记GFP(g)嘌呤霉素(h)GFP和嘌呤霉素(i)
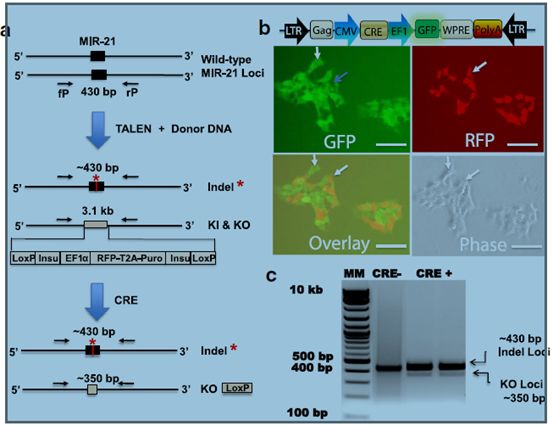
图2.为检测Cre功能构建TALEN-工程化的HEK293报告基因的方法。a.示意图展示了未经修饰的HEK293细胞在两个loci(上边条带)都有野生型的MIR-21基因。TALEN的引入和HR供体DNA导致MIR-21双等位基因的消失(中间条带)。这些TALEN-工程化的HEK293细胞(HEK293-TE)可以作为检测CRE活性的功能性使用的报告细胞系(下边条带)b.转染慢病毒-Cre-GFP载体到HEK293-TE细胞中。绿色荧光表明了Cre-GFP的表达(左上),红色荧光表明了floxed-RFP-Puro的表达(右上)。覆盖区域表明表达Cre-GFP的细胞不和floxed-RFP-Puro细胞重叠(左下)。c.引入Cre-GFP后对HEK-293细胞进行基因分型。上边约430bp的是突变等位基因,下边约350bp条带和移除floxed报告基因片段的小PCR产物相同。floxed报告基因片段(3.1kb)等位基因不能在我们的实验条件下扩增。白色箭头表明GFP和RFP阳性相互排斥。比例尺50μm。
靶向核酸酶已被成功用于癌症研究、进化研究和基因治疗中的基因敲入和敲出,并能通过它们强健的核酸酶活性产生双等位基因改变。我们之前用TALEN配对和HEK293细胞中的同源重组载体完成了双等位基因MIR-21的基因敲除(图2a)。使用构建的TALEN-设计的HEK293细胞(HEK293-TE),在这项研究中,我们为我们新设计和构建的Cre表达载体设计了一种监测Cre活性的方法。因为这个方法清楚地证明了将靶向核酸酶技术与Cre-LoxP和病毒转导载体结合以完成更先进的基因组编辑的效果,这个方法很严谨且与我们的研究高度相关。
为了检测这个方法的可行性,我们用表达Cre和GFP的慢病毒载体转染了红色荧光HEK293-TE细胞(图2b,右上)。基因组分型和序列分析表明一个HEK293E克隆中一个等位基因上有一处插入缺失标记,另一个等位基因上有基因敲入和基因敲除改变(图2a 中上;图c)。通过肉眼观察,转染后第一天一部分细胞出现绿色荧光,说明瞬时转染后转基因(Cre)迅速表达。第二天,绿色荧光增强,而红色荧光减弱,表明工程细胞里侧面含有RFP-嘌呤霉素的基因被敲除。第三天,与RFP荧光信号相比,表达Cre-GFP的细胞更加明显,如图2b所示(左上)。红色和绿色荧光不在同一区域重叠,说明这些细胞表达侧面含有RFP-嘌呤霉素基因或Cre-GFP(图2b,下)。我们将双向排斥现象归结于Cre基因的成功地转导和稳健的表达以及随后侧面含有RFP-嘌呤霉素序列的敲除。这个转染数据表明了Cre-LoxP技术的有效性。
随后,我们通过基因分型分析证明了在HEK293-TE报告细胞中移除floxed RFP-嘌呤霉素基因。在TALEN-和Cre-工程化细胞中,插入缺失标记位点(~430bp)均一致出现,表明了NHEJ修正的等位基因的PCR产物。在我们的PCR条件下,基因敲入等位基因通过限制PCR延伸时间不扩增。因此,TALEN-工程化的细胞有一个PCR产物,而Cre-工程化的细胞有两个PCR产物,包括插入删除标记(~430bp)和成功的移除敲入的floxed RFP-嘌呤霉素片段(~350bp)。正如预测的一样,只有在细胞中引入Cre(Cre+条带)后才出现敲除位点(KO,~350bp)和移除的floxed-RFP-嘌呤霉素片段区域(图2c).这些结果证明了Cre在工程细胞中敲除了floxed序列。此外,荧光变化提供了一种实时监测哺乳动物细胞中Cre活性的方便的方法。因此,我们用显微镜和这些报告细胞的荧光特征完成我们剩余的研究。
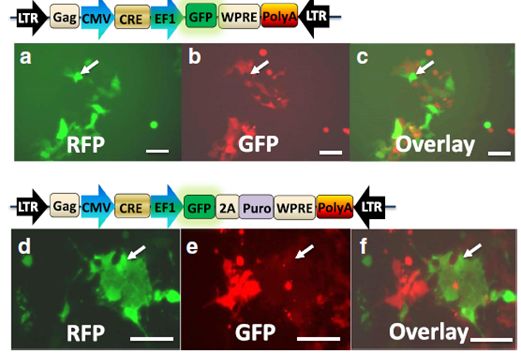
图3.用慢病毒转导的方法有效地移除foxed报告基因。通过病毒转导功能性地验证表达Cre的载体。HEK293-TE细胞丰度长到20~40%时,用慢病毒-Cre-GFP(a-c)或慢病毒-Cre-GFP-Puro(d-e)转染5天,并在20倍照相。绿色荧光表明Cre-GFP或Cre-GFP-Puro的表达(a-d)。红色荧光表明floxed-RFP-puro的表达(b,e).分别覆盖A/B或D/E表明表达Cre-GFP的细胞不和floxed-RFP(c)或floxed-RFP-puro(f)细胞重叠。白色箭头表明GFP和RFP阳性相互排斥。比例尺为50μm。
慢病毒-Cre高效地移除HEK293-TE 报告细胞floxed RFP-嘌呤霉素基因
之前,我们和其他科研人员证明了水疱性胃病毒包膜糖蛋白(VSVG)-假型慢病毒具有超高的感染率~100%。在这个研究中,我们选择VSVG假型产生慢Cre病毒,并检测VSVG假型产生的慢Cre-GFP能否在报告细胞中转导、表达并随后编辑敲入的floxed-RFP-Puro基因片段。我们用低病毒复数0.5感染HEK293报告细胞检测慢病毒体系的有效性和稳健性。如预测的一样,在病毒感染后的第二天我们开始观测到一个弱的转基因GFP表达。第三天时GMP信号变强且明显,表明与上述转染方法相比,此方法转基因表达相对较慢。同时,在GFP阳性细胞中,RFP信号变弱。第5~7天GFP和RFP的荧光信号都变强和明显(图3 a-c)。在这些慢病毒感染数据中,GFP和RFP荧光相互排斥说明了慢病毒Cre转染体系基因编辑的稳健性和有效性。在相同的实验条件下,另一个慢病毒Cre-GFP-Puro载体在移除floxed RFP-Puro基因序列中以相似的方式表现出相同的活性(图3 d-f)。这些结果得出两个重要的结论:一、慢病毒转导受体细胞是稳健的;二、与转染方法相比,Cre编辑的发生推迟了一些。推迟的时间很可能是慢病毒Cre需要合成宿主基因组的时间。然而,在转染中,Cre不需要合成所以立即开始表达。总之,转染方法的转染速率较慢但能迅速开始转基因的表达,这很适合易转的细胞类型,而转导方法转染速率较快但起始较慢,这更适合难转染的细胞类型。此外,利用可选择的标志物,我们的慢病毒Cre方法都能用于荧光激活细胞分离仪(FACS)和简单的药物筛选。并且,我们的慢病毒-Cre体系为哺乳动物更先进的基因组编辑技术提供了稳健、灵活的方法。
AAV-Cre在HEK293-TE报告细胞中通过转染和转导方法均能移除floxedRFP基因
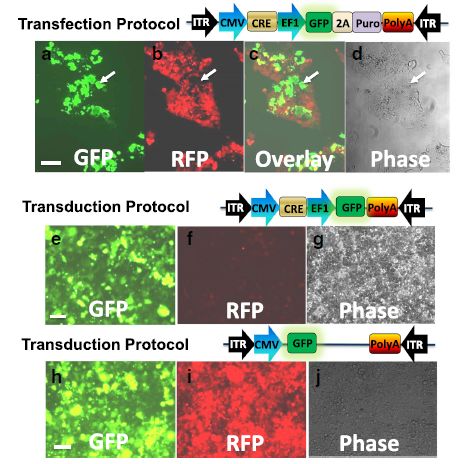
图4.在工程化的HEK293细胞中进行以AAV为基础的Cre功能性验证。用表达Cre和GFP的构建物转染HEK293-TE细胞(a-d)。图像为转导后3天,20倍放大拍摄。绿色荧光指代表达Cre-GFP-puro的细胞(a)。红色荧光指代表达floxed-RFP-puro的细胞(b)。分别重叠左边和中间的图像说明表达细胞表达RFP或GFP(c),和重叠的图像(d)。或者,以表达Cre-GFP(e-g)或对照GFP病毒(h-j)2.0MOI的AAV转导20%~30%丰度的细胞。转导后10天,20倍放大下拍摄。绿色荧光表明表达Cre-GFP(e)或对照GFP(h)。消失的红色荧光表明floxed-RFP-puro的表达(f)。对比之下,在GFP非编辑对照中红色荧光信号仍然很强(i)。和对照细胞(j)相对比表明TALEN-Cre序列性地编辑HEK293细胞(g)。白色箭头表明GFP和RFP阳性相互排斥。比例尺为50μm。
与整合慢病毒相比,AAV可以不整合宿主细胞基因组将基因转导至细胞。随后我们研究了HEK293-TE报告细胞中AAV-Cre基因组编辑活性。首先,我们验证了AAV载体能否通过简单的转染方法使用。如图4所示,转染AAV-Cre-Puro后,一部分细胞出现了GFP(图 4a),同时在相同的细胞中RFP消失(图 4b-d),说明通过向工程细胞中引入Cre基因成功地移除了floxed RFP-Puro基因片段。
我们在AAV-Cre转染方法中的成功鼓励我们在HEK293-TE细胞中进一步检测转导方法的效果。尽管AAV技术用于体内,在体外细胞培养条件下AAV的感染效果不如慢病毒,感染率也较低。然而,因为AAV的低毒性,我们用高病毒复数的病毒(2MOI;使用Cre-GFP编辑或者GFP非编辑对照病毒)转导报告细胞。在细胞培养条件下,感染效果较弱,转基因GFP在第3天开始出现第4~5天增强(图 4e-j)。然而,转导后,RFP在第6~7天开始消失(没有数据图)。第10天,几乎所有Cre组的细胞都变成了GFP阳性和RFP阴性(图4 e-g),表明通过转导的Cre GFP编辑病毒从HEK293-TE细胞中成功地移除floxed RFP-Puro基因片段。与之相比,在GFP非编辑对照组里细胞变成了GFP阳性(图 4h)但仍然保持RFP阳性(图 j)。综上,我们使用易操作的转染和病毒转导方法验证了AAC-Cre转导体系。AAV的非整合特性使得它非常适合临床应用。









