吉非替尼的两种核磁共振定量方法
Determination of Gefitinib by Two Methods of Quantitative Nuclear Magnetic Resonance
郝海军,贾幼智,韩 茹*,胡 彬
(中国医药工业研究总院上海医药工业研究院,上海 201203)
摘要:分别采用氢核磁共振定量法和氟核磁共振定量法测定吉非替尼含量。氢核磁共振定量法以化学位移δ7.21 为定量峰,马来酸δ6.02 为内标峰,在测试温度300 K,谱宽8 012 Hz,采样时间4.01 s,弛豫延迟时间20 s,脉冲宽度9.54 μs,扫描次数64 次条件下采集氢谱。氟核磁共振定量法以4- 溴-2- 氟乙酰苯胺为内标,脉冲序列zgfhigqn.2 在恒温300 K 下采集氟谱。两种核磁定量方法都进行了方法学验证。吉非替尼氢核磁共振定量法和氟核磁共振定量法含量测定结果与质量平衡法结果基本一致。本研究建立的核磁共振定量方法专属性强、快速、准确,可用于吉非替尼的含量测定。
关键词:吉非替尼;氢核磁共振定量法;氟核磁共振定量法;含量测定
吉非替尼( gefitinib,1),是一种表皮生长因子受体(EGFR) 酪氨酸酶抑制剂,临床主要用于治疗局部晚期或转移性非小细胞肺癌[1—2]。1 含量测定方法主要有紫外分光光度法、高效液相色谱法和液质联用色谱法等[3]。采用核磁共振定量法(quantitative nuclear magnetic resonance,qNMR) 测定1 含量尚未见报道。
随着高强度磁场及傅里叶变换技术的应用,核磁仪器的性能得到极大改善,从而为核磁共振技术在定量分析领域的应用奠定了基础。中国、美国及英国药典均收载了qNMR 法[4]。qNMR 法主要有氢核磁共振定量法( qHNMR) 和氟核磁共振定量法( qFNMR)。质量平衡法作为一种基准方法,被世界卫生组织(WHO) 和欧洲药典委员会推荐为药品标准物质定值方法。本研究分别采用qHNMR 和qFNMR 法测定1 含量,并与质量平衡法结果比较,为1 含量测定及标准品标定提供新的方法,也为qNMR 法在定量方面的应用提供参考。
1 仪器与试药
2 方法与结果
2.1 核磁定性分析结果
2.2 qHNMR 法测定1 含量
2.2.1 qHNMR 核磁条件
采用zg30 脉冲序列获取1H NMR 图谱。参数:测定温度 300 K,谱宽(SWH)8 012 Hz,中心频率(O1)2 648.5 Hz,采样时间(AQ)4.01 s,弛豫延迟时间(D1) 20 s,脉冲宽度(P1) 9.54 μs,空扫次数(DS)2 次,扫描次数(NS)64 次。
2.2.2 供试品溶液的配制
2.2.3 专属性考察
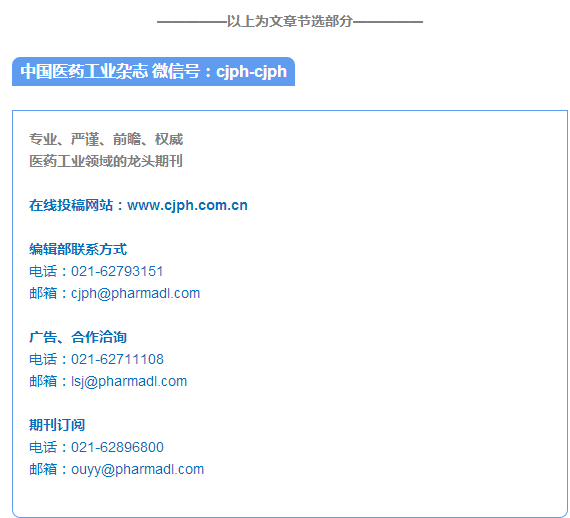
按“2.2.1”项下条件,设置仪器参数、调谐、匀场和采集图谱,调整相位及基线。样品1 与内标2 混合溶液的1H NMR 图谱见图1。δ3.94 处的-OCH3 峰含有3 个质子峰,峰面积较大,较适合作为定量峰。但由于受到附近水峰的影响,导致该处基线不平。结果表明,1 的质子信号δ7.21 作为定量峰,2 的质子信号峰δ6.02 为内标峰时,能够完全分离,互不干扰,基线平坦,专属性较高。
2.2.4 线性及范围
用2 内标溶液分别配制系列浓度1 溶液(1.0 ~10.0 mg/ml)。混匀后分别转移0.6 ml 至5 mm 核磁管中,待测。以1 与2 定量峰面积比x 为横坐标,1 与2 质量比y 为纵坐标,进行线性回归,得回归方程y=2.251 1x-0.097 3(r=0.999 3)。在1.0 ~10.0 mg/ml 范围内,1 的定量峰面积与其质量的线性关系良好。
2.2.5 精密度、重复性和稳定性试验
取1 供试品溶液,按“2.2.1”项下条件连续测定6 次,计算1 定量峰与内标峰面积的比值,结果RSD 为0.24%。
按照“2.2.2”项下方法配制6 份供试品溶液,分别进样,计算1 含量,结果RSD 为0.28%。取1 供试品溶液,分别于0、4、8 和12 h 按“2.2.1”项下条件进样测定,计算定量峰与内标峰面积比值,结果RSD 为0.96%,表明样品溶液于室温放置12 h 内稳定。
2.2.6 含量测定
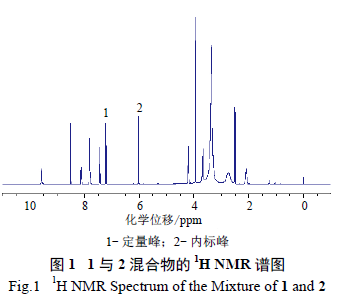
分别精密称取3 批1 原料药,按“2.2.2”项下方法配制供试品溶液,分别进样测定,调整相位并积分,按内标法( 下式) 计算1 含量。结果1 含量分别为91.74%、90.90%和91.61%。
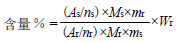
式中:As 为1 定量峰的积分面积;ns 为1 定量峰代表氢原子数;Ms 为1 相对分子质量(Mr);ms 为1质量;Ar 为2 内标峰的积分面积;nr 为2 内标峰所代表氢原子数;Mr 代表2 的Mr;mr 代表2 质量;Wr 为内标2 的百分含量。
2.3 qFNMR 测定1 含量
2.3.1 qFNMR 核磁条件
采用zgfhigqn.2 脉冲序列在恒温(300 K) 条件下获取19F NMR 图谱。参数设置如下:谱宽(SW)δ10,射频中心频率(O1P)δ-122.6,采集点数(TD)64K。采样时间(AQ)4.02 s,弛豫时间(D1)10 s,采样次数 32 次,空扫次数(DS)4 次,增益(RG)203。
.3.2 供试品溶液的配制
精密称取3 25.0 mg,用DMSO-d6 定容至25 ml,作为内标溶液。精密称取1 原料药10.83 mg,加入3 内标溶液1.0 ml 溶解,混匀后转移0.6 ml 至5 mm核磁管中,待测。
2.3.3 专属性考察
按“2.3.1” 项下条件, 设置仪器参数、调谐、匀场和采集图谱,调整相位及基线,1 与内标物的19F NMR 图谱见图2。与1H NMR 图谱相比,19F NMR 图谱简单、直观。1 的19F NMR 图谱在δ-121.69 相应峰与内标物在δ-123.31 相应峰分离良好,因此选择δ-121.69 作为定量峰。
2.3.4 线性与范围
2.3.5 精密度、重复性和稳定性试验
取1 供试品溶液,按“2.3.1”项下条件连续测定6 次,计算1 定量峰与3 内标峰面积的比值,结果RSD 为0.34%。
按“2.3.2”项下方法配制6 份供试品溶液,计算1 定量峰与3 内标峰面积的比值,结果RSD 为0.58%。
取1 供试品溶液,分别于0、4、8 和12 h 按“2.3.1”项下条件测定,计算1 定量峰与3 内标峰面积比值,结果RSD 为1.75%,表明供试品溶液于室温放置12 h 内稳定。
2.3.6 定量限(LOQ)
据报道[6],19F NMR 用于定量分析时,信噪比(S/N) 须大于300。据此确定本法LOQ 为3.44 mg。
2.3.7 含量测定
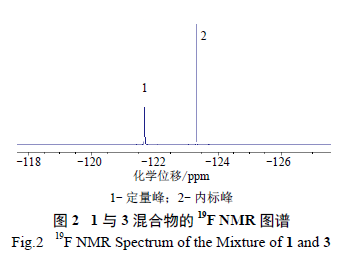
分别称取3 批1 原料药,按“2.3.2”项下方法配制供试品溶液,进样测定,调整相位并积分。按照下式计算1 含量。
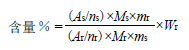
式中:As 为1 定量峰的积分面积;ns 为1 定量峰代表氟原子数;Ms 为1 分子量;ms 为1 质量;Ar 为3 内标峰的积分面积;nr 为3 内标峰所代表氟原子数;Mr 代表3 分子量;mr 代表3 质量;Wr 为内标3 的百分含量。结果1 含量分别为91.08 %、90.89 % 和91.18%。
2.4 与质量平衡法的比较
为验证qHNMR 和qFNMR 法测定结果的准确性,采用质量平衡法对3 批1 原料药进行了含量测定[7]。
3 讨论
19F 的天然丰度为100%,磁矩与1H 的磁矩接近,19F NMR 的灵敏度也与1H NMR 接近,因此比较适用于qNMR。与1H NMR 相比其最大优势是,19F 图谱范围一般为δ- 200 ~ -100,且响应峰个数很少,因此不会发生响应峰重叠现象;而1H NMR图谱范围一般为δ0 ~ 16,响应峰很容易发生重叠,往往影响定量峰的选择。
在优化qHNMR 测试参数时发现,各个参数的变化趋势有较大差别。但不论何种变化趋势,确定测试参数时应选择定量峰与内标峰面积比值稳定时的初始值[8]。内标峰与定量峰的射频场不同时会影响试验结果的准确性,因此需要对射频中心频率(O1P) 的位置进行调整,使之位于内标峰与定量峰的中间位置。本试验最终确定O1P 为6.62 ppm,对应O1 值为2 648.5 Hz。
确定qFNMR 测试参数时也须采用定量峰与内标峰面积比值稳定时的初始值。另外,19F NMR 用于定量研究时需要先测定图谱的谱宽(SW),然后适当缩小SW,并调整O1P 位置,使之位于定量峰与内标峰的中间位置。这样可以方便相位校正,保证定量分析结果的准确性。本研究在预试验的基础上,调整SW 为10,O1P 为δ-122.6。
经验证, 本研究建立的qHNMR 和qFNMR法测定结果与质量平衡法测定结果基本一致,而qNMR 用于含量测定无需对照品,样品消耗量小,操作简单,结构鉴定与含量测定可同步完成,与质量平衡法相比有其独特优势。
作者简介:郝海军(1981—),男,硕士,助理研究员,从事新型给药系统及
仪器分析研究。
Tel:021-20572000×1041
E-mail:[email protected]
通信联系人:韩 茹(1978—),女,副研究员,从事药物分析研究。
Tel:021-20572000×4058
E-mail:[email protected]