
今天应该是本系列文章的第三篇了,不知大家有没有视觉疲劳?咳咳,当然了,本宫也就是礼貌性地问一问,此类文章还有许多,写还是会写的。
今天的文章是"
MicroRNA
and
Transcription Factor
Gene Regulatory
Network Analysis
Reveals Key Regulatory Elements Associated with
Prostate Cancer
Progression",发表在
PLOS ONE
上(此杂志本宫就不多作介绍了),作者都是歪国仁,单位以伊朗的为主,还有印度的和土澳的,估计是伊朗的人去那俩地方留学,别问本宫为何知道的辣么多~
言归正传,本文研究的是
前列腺癌
,用的样本是
对应的非癌样本
以及
转移
和
非转移
3组样本,主要方法是构建
miRNA、mRNA与转录因子(TF)的调控网络
,来揭示比较癌症转移和非转移组中的Keg基因。
下面介绍一下文章的主要思路和方法
1、GEO数据集获取和差异基因筛选
这也算是老套路了,作者选取的数据集是GSE21032,这个数据集是个很大的数据集,作者从218个样本中挑选出
139个miRNA和mRNA均有数据的样本
进行分析。差异基因的筛选标准也是P<0.05和log(FC)的绝对值>1,这部分可以直接用
GEO2R分析
再用
EXCEL简单处理
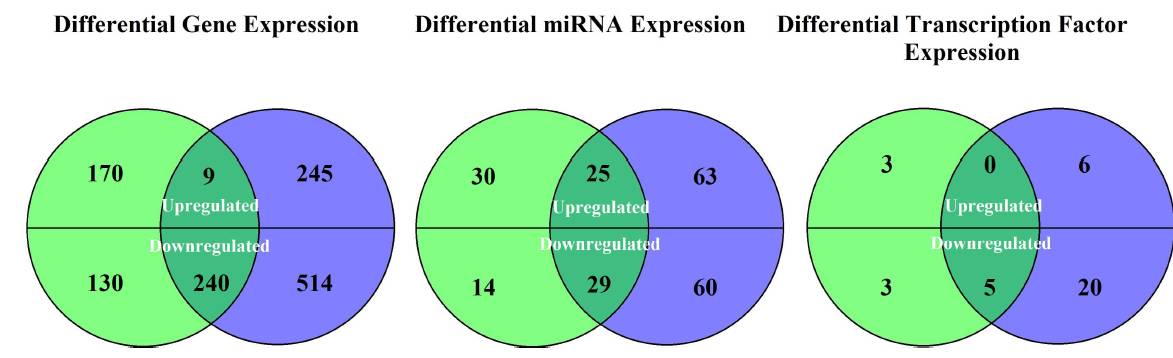
即可,小张之前也介绍过。最终,作者在原发癌组、转移组中分别筛选出下图所示的差异miRNA、mRNA以及TF,当中有一些有交集,有一些没有完全没有交集(比如上调的TF)。
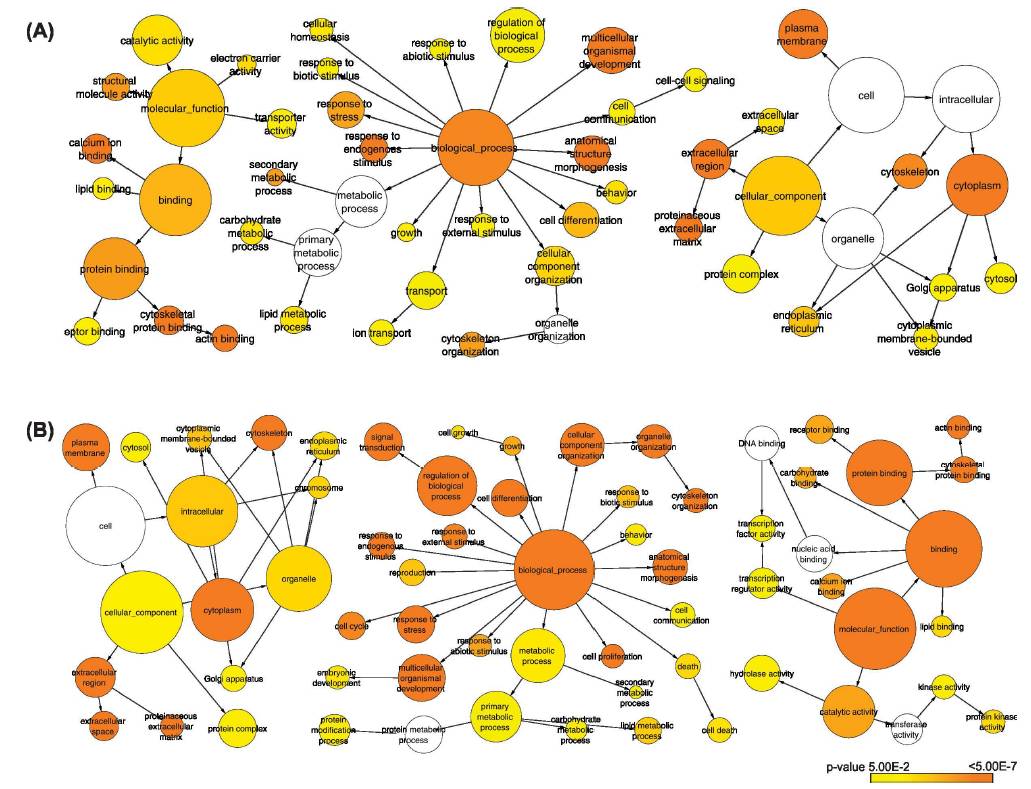
2、生物学功能分析
作者对差异mRNA进行了GO分析,利用的工具是
Cytoscape的插件BiNGO
。通过下列的网络图简单地阐述了疾病与相关生物学功能的关系。
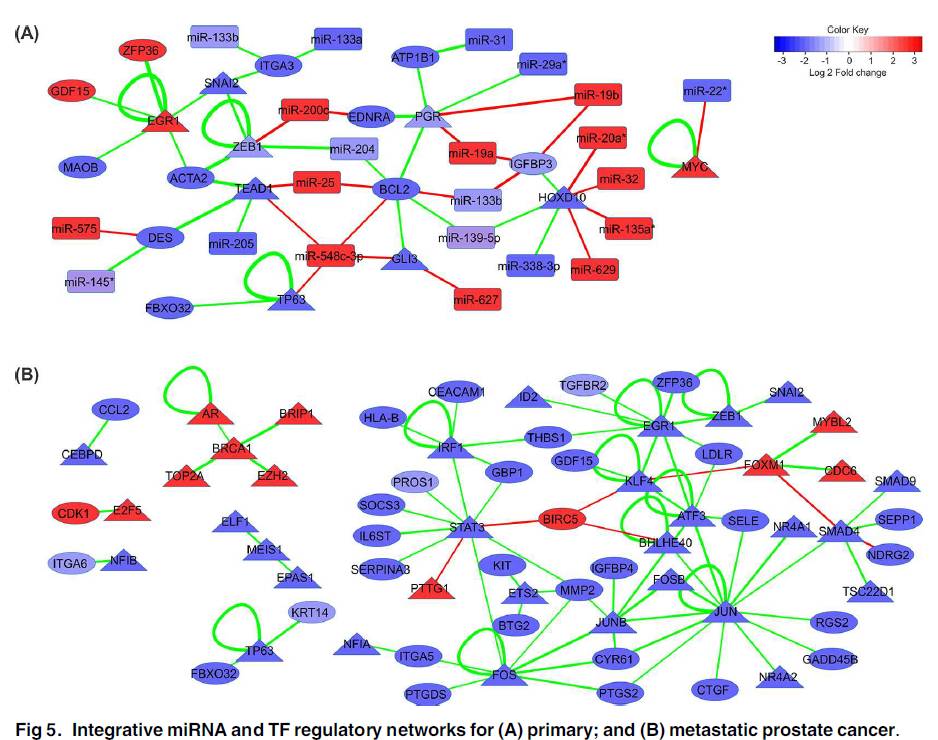
3、TF调控网络分析与miRNA调控网络分析
TF与基因的关系通过
TRANSFAC数据库
获取,这个数据库是TF数据库中公认的比较好用的数据库,然而不是一个完全免费的数据库,免费使用只能使用截止到05年的数据,如果
在高校中有条件
的童鞋,可以试试这个数据库。获取了TF-mRNA配对的相关数据之后,再在芯片数据中
分析TF与mRNA的相关性
(利用
皮尔森相关性系数
)。miRNA与基因的关系如何预测想必大家应该比较熟悉了,之前的文章中已经介绍了许多miRNA相关的数据库,作者用了
MirTarbase、miRanda和TargetScan
三个数据库来预测miRNA与mRNA的作用关系。同样的,作者利用表达相关性进行进一步筛选以降低假阳性率。
网络构建好了,key基因如何筛选?本文中作者主要应用了两个网络拓扑结构参数,一个就是之前常说的
degree
,另一个是
中介中心性(Betweeness Centrality)
,这是一个网络中地位重要性的图论概念,简单的说就是
中心性越高表示该节点在网络中的重要性越高
。这两个参数利用
Cytoscape的CentiScape
插件都可以轻松搞定。
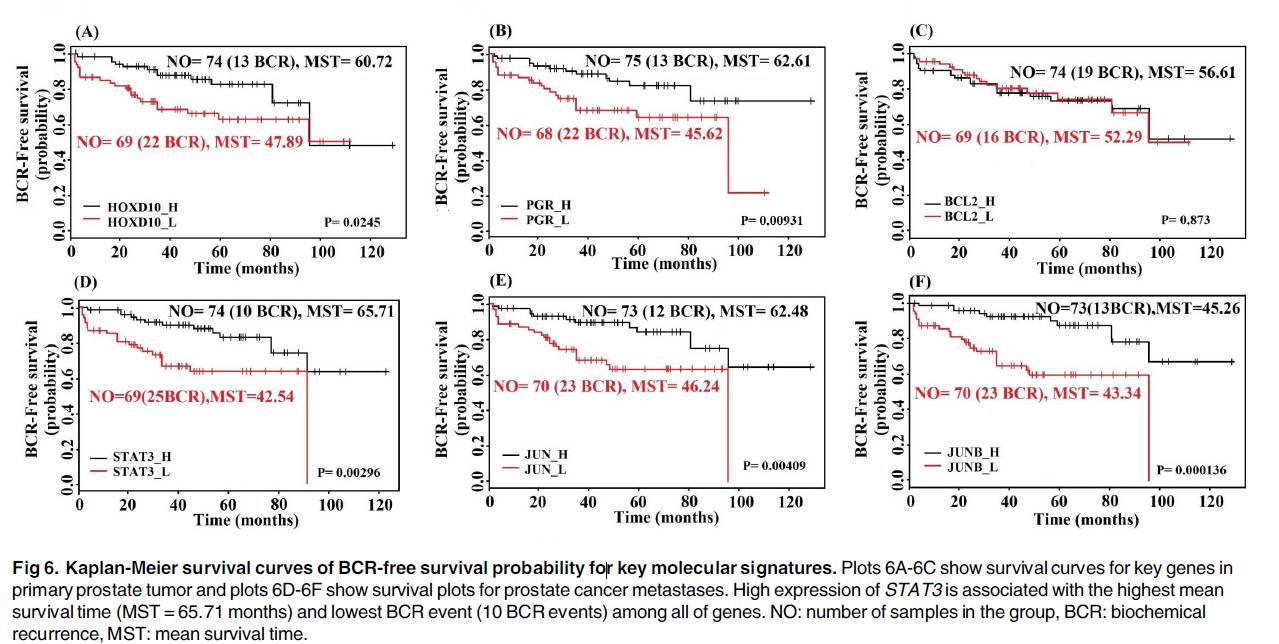
4、生存分析
最终作者筛选出两个关键的转录因子HOXD10和PGR,并运用生存分析进行验证,结果表明这两个转录因子确实在前列腺癌的转移过程中起到了重要作用。
小结:
利用数据库或者软件预测出来的结果往往会出现假阳性,比如说TRANSFAC数据库对转录因子的预测,是基于
Bind Sites
的预测,Bind Sites位于Gene的调控区内,是很短的一段序列,与转录因子结合,调控基因转录,
由于Bind Sites本身太短,所以特异性较差
,预测出来的结果可能很多都是假阳性的,所以我们要通过进一步的筛选,比如文章中用的基因表达的
相关性分析
,
网络拓扑结构分析
等等来降低假阳性率,最后再通过生存分析验证,让整个文章的结果有理有据让人信服。其它的数据库比如miRNA靶基因的预测,我们往往需要用
多个数据库的结果取交集
,也同样是为了提高数据的可信度。










