
▲第一作者:葛瑶;
通讯作者:丁奎岭,韩召斌;
通讯单位:中国科学院上海有机化学研究所
论文DOI:10.1021/jacs.9b02920
全文速览
本研究将手性 Ir(SpinPHOX)(COD)BAr
F
络合物成功应用于 3,6-双亚烷基-2,5-二酮哌嗪衍生物的双不对称氢化反应,以优异的产率(94- > 99% yields)和对映选择性(up to 98% ee)及立体专一的
cis
选择性 (>99/1
de
)得到了一系列手性环二肽类化合物。利用这一方法完成了三个天然产物,一个桥环化合物以及一个单边羟基化的哌嗪衍生物的合成。对氢化反应机理的系统研究表明,氢化反应可能主要通过催化剂“非解离”的模式连续进行的,即底物分子中一个双键首先被铱催化剂还原生成的单边氢化产物依然与催化剂的中心金属结合,催化剂在底物分子中通过分子内的“转移”后,继续对第二个双键进行氢化生成最终产物。这是一种罕见的催化剂“非解离”方式氢化含有两个双键的杂环化合物的例子,可谓一箭双雕。
背景介绍
含有 3,6-二取代-2,5-二酮哌嗪这一核心骨架的手性环二肽大多具有独特的生物活性,近年来在药物分子的设计和研发中备受关注。此外,手性 3,6-二取代-2,5-二酮哌嗪衍生物还可以作为有机小分子催化剂或手性辅基参与不对称合成反应。合成 3,6-二取代-2,5-二酮哌嗪化合物的经典方法是将氨基被保护的氨基酸与氨基酸酯缩合,再通过脱除保护基后进行原位环化(图1, a)。虽然该方法具有一定的通用性,然而需要谨慎选择反应条件(如 pH 值)以避免过程中可能出现的手性中心的部分消旋化。1990年,Takeuchi 等人在一种哌嗪生物碱的合成中尝试了钴催化的 3,6-二亚苄基-2,5-二酮哌嗪的不对称氢化。然而,该反应仅取得了 40 % 的收率和中等的对映选择性(82 % ee, 图1, b)。鉴于手性 3,6-二取代-2,5-二酮哌嗪衍生物(环二肽)潜在的应用价值,采用催化方法高效高选择性构建该类结构依然是一个值得深入探索的问题。
研究出发点
课题组在前期研究工作中发展了一类基于手性螺[4,4]-1,6-壬二烯骨架的膦-噁唑啉配体 SpinPHOX 及其阳离子型铱络合物 Ir(SpinPHOX)(COD)BAr
F
。这一铱催化体系可以高效、高选择性地实现亚胺和多种共轭双键的不对称氢化
(
Angew. Chem., Int. Ed.
2009
,
48
, 5345;
Chem. Commun.
2010
,
46
, 156;
Chem. Commun.
2012
,
48
, 5172;
Angew. Chem., Int. Ed
.
2012
,
51
, 936;
Angew. Chem., Int. Ed.
2014
,
53
, 1978;
Angew. Chem., Int. Ed.
2018
,
57
, 13140)
。在这些前期工作的基础上,作者认为通过合理的对 Ir/SpinPHOX 催化体系进行优化,有望通过不对称催化氢化的方法高效高选择性地制备 3,6-二取代-2,5-二酮哌嗪(环二肽)类化合物(图1, c)。
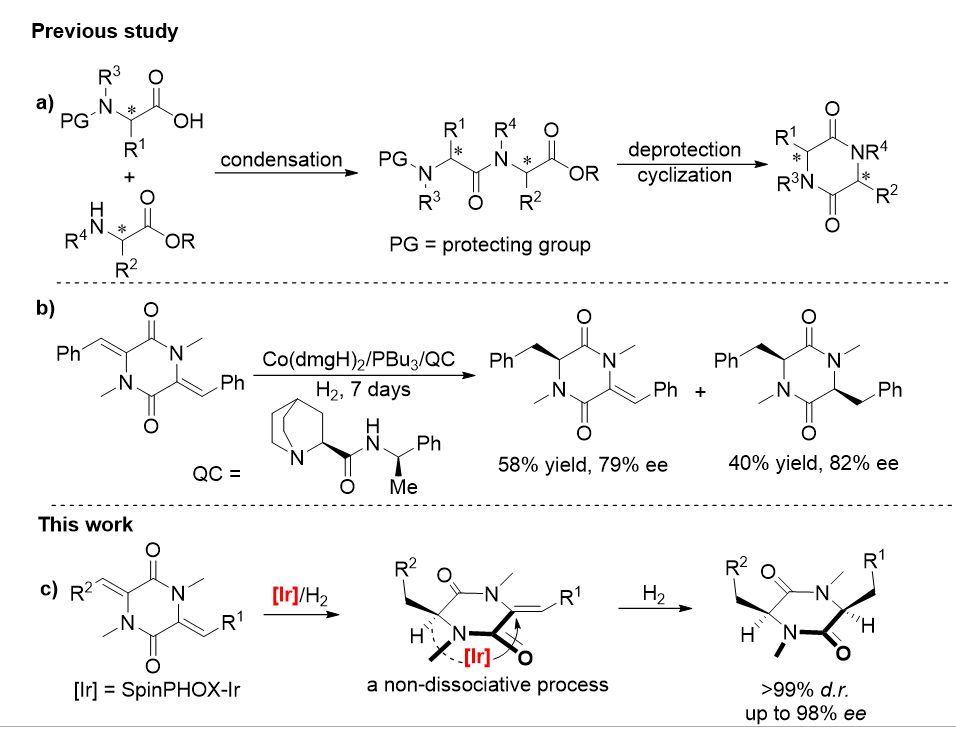
▲图1. 一些 2,5-二酮哌嗪衍生物的合成方法及本文的工作
图文解析
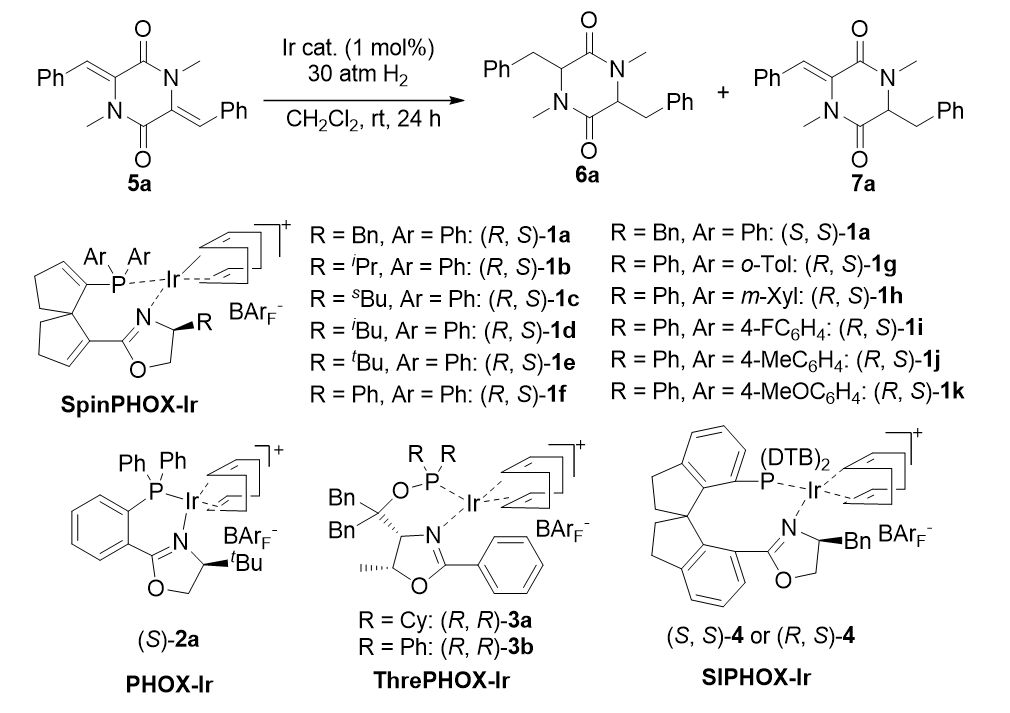
▲图2. 对催化剂的考察
通过对一系列手性铱络合 Ir(SpinPHOX)(COD)BAr
F
在标准底物
5a
的催化氢化反应中的效果考察(图2),最终确定 SpinPHOX/Ir-(
R
,
S
)-
1k
为最适合该反应体系的催化剂。一些具有优势结构的铱催化剂如 PHOX-Ir (
S
)-
2a
,ThrePHOX-Ir (
R
,
R
)-
3a
,(
R
,
R
)-
3b
以及SIPHOX-Ir (
S
,
S
)-
4
,(
R
,
S
)-
4
在该反应中并未表现出明显的反应活性和/或手性控制能力。
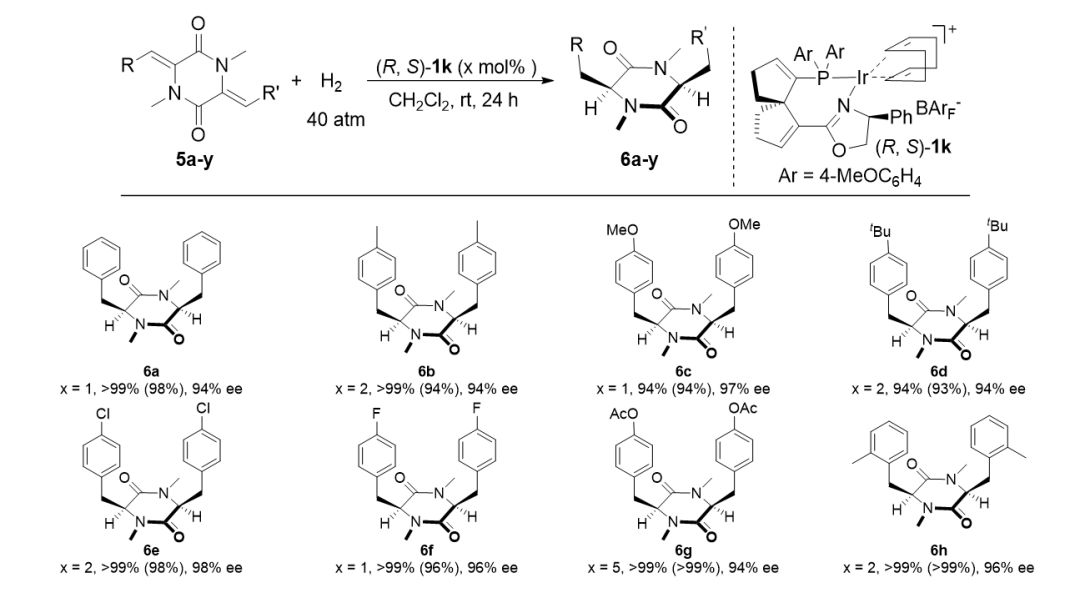
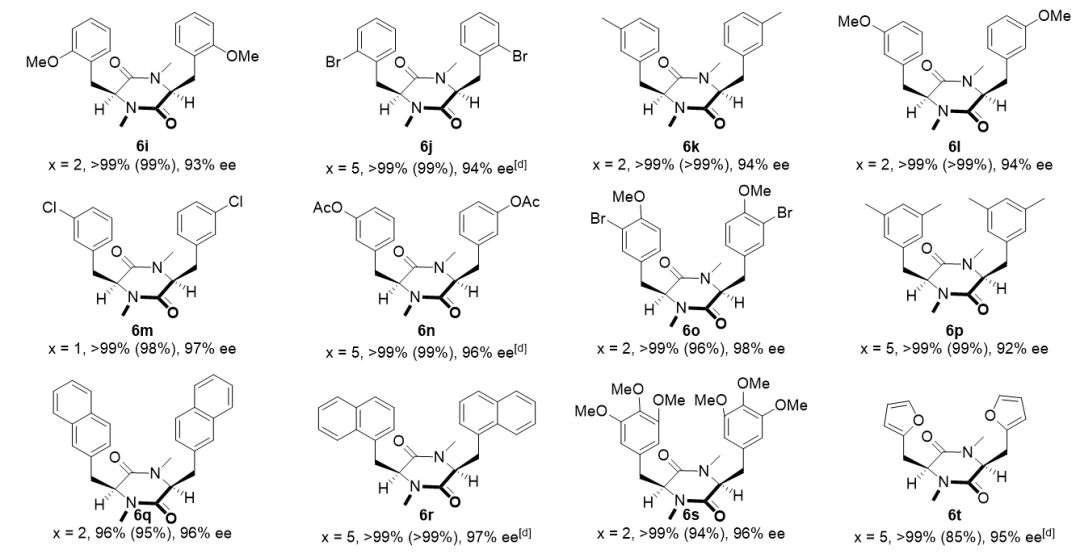
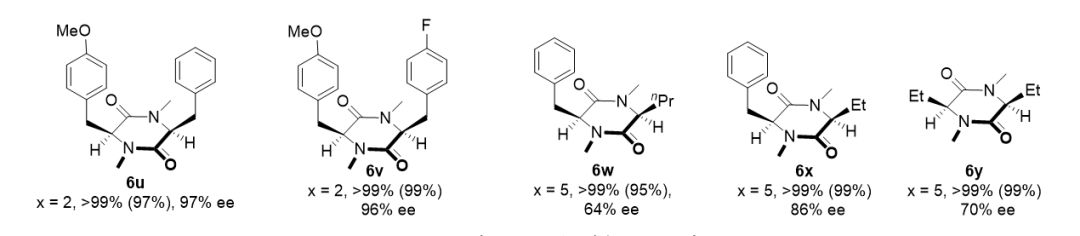
▲图3. 底物适用性的考察
在优化的反应条件下,一系列 3,6-二亚烷基-2,5-二酮哌嗪衍生物可以顺利的进行氢化,以良好到优秀的对映选择性,专一的顺式选择性生成手性环二肽化合物。催化体系具有良好的官能团兼容性,对于含芳环、杂环及烷基取代基的底物都表现出了良好的适用性。(图3)
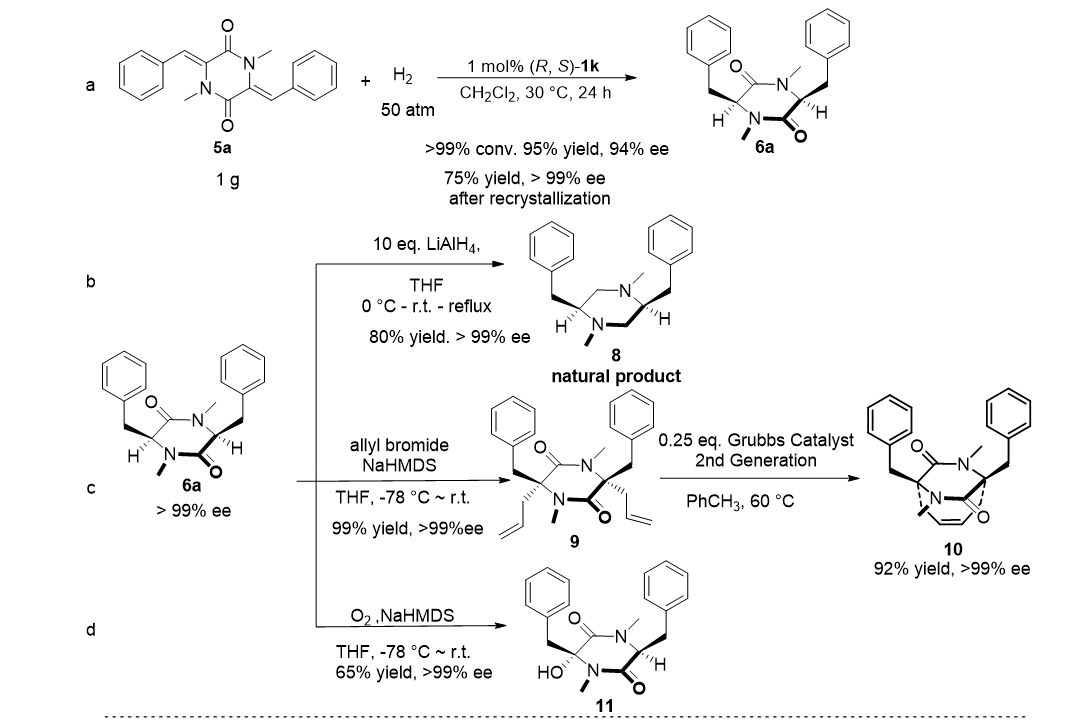
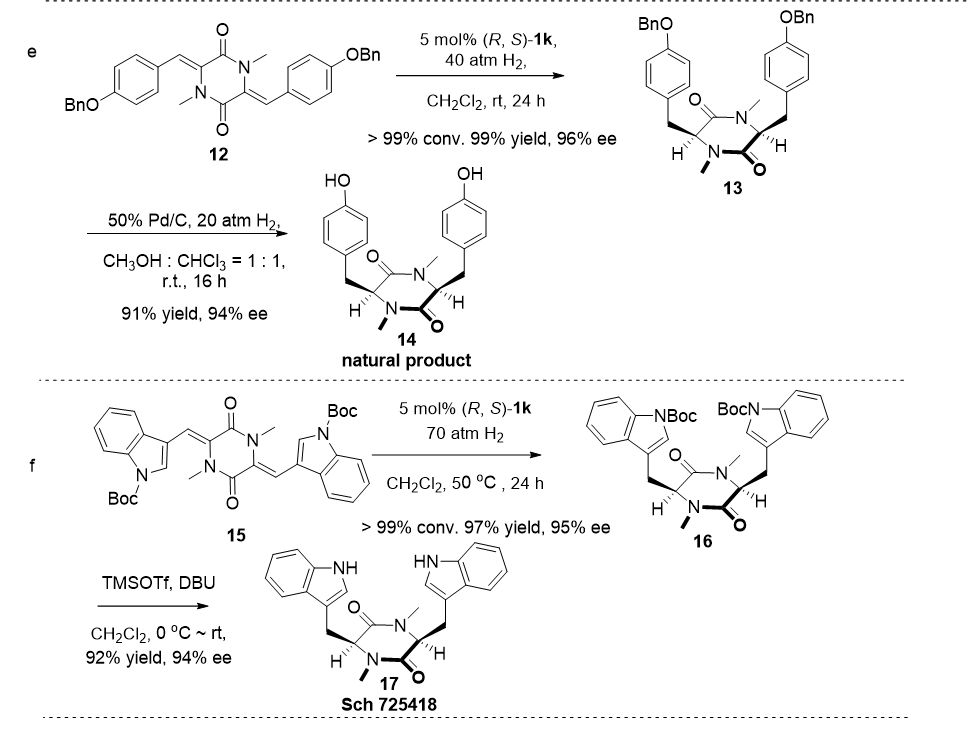
▲图4. 氢化产物的衍生化研究
对标准底物
5a
的不对称催化氢化可顺利放大至克级规模,产物(
S
,
S
)-
6a
的光学纯度通过一次重结晶可提升至 > 99 % ee (图4a )。(
S
,
S
)-
6a
可方便地转化为天然产物
8
、桥环化合物
10
以及羟基取代的哌嗪衍生物
11
(图4b-d )。此外,以(
R
,
S
)-
1k
催化的
12
和
15
的不对称氢化为关键步骤,合成了天然产物
14
,
17
(图 4e-f )。
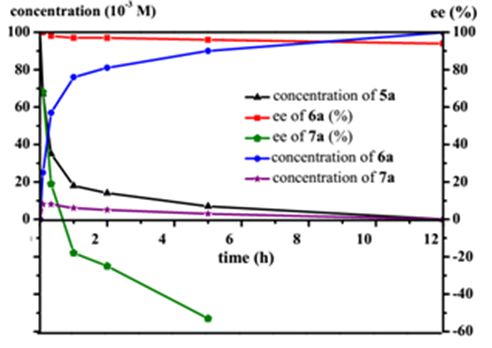
▲图5. 对
5a
不同反应时间氢化反应的监测
以
5a
为底物,对氢化反应的机理进行了探索。对反应进程的监测(图5)表明,在整个反应过程中两个双键均被还原的顺式产物(
S
,
S
)-
6a
始终是主要产物,其ee值随产率的提升由 >99 % 相应下降至 94 %。单边还原产物
7a
的产率始终保持在较低的水平(<8%),且随着反应的进行其绝对构型由
S
翻转为
R
。因此可以推测:(
R
,
S
)-
1k
在对光学活性的
7a
进行催化氢化时,出现了动力学拆分的作用。(
R
,
S
)-
1k
对(
S
)-
7a
的氢化更为有利,进而造成了(
R
)-
7a
在体系内的富集。
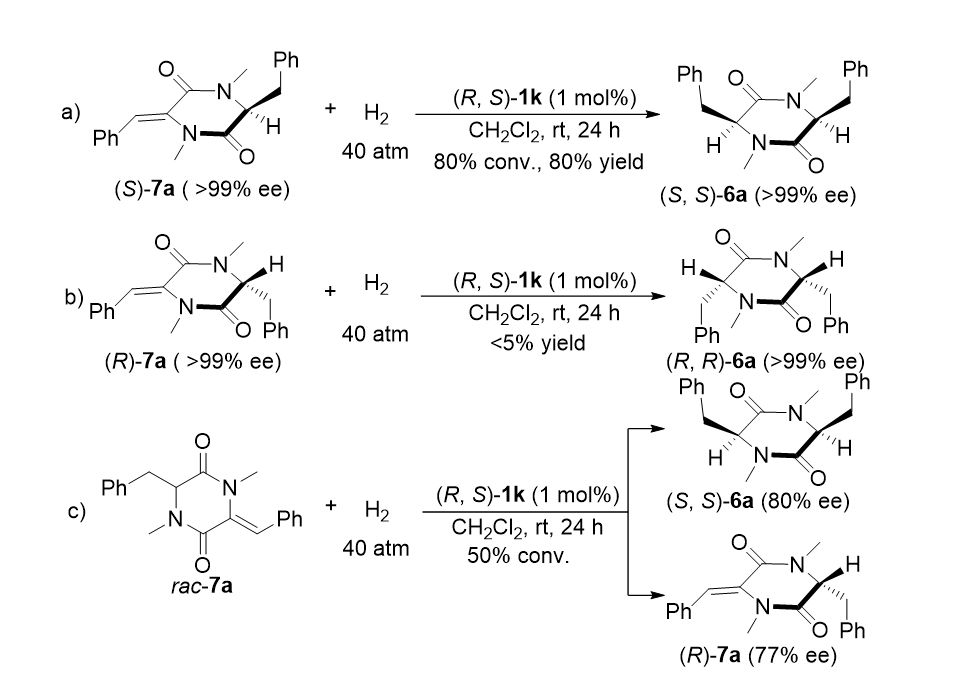
▲图6. 动力学拆分现象的实验验证
为了确认动力学拆分现象的存在,我们进一步进行了控制实验(图6)。光学纯的(
S
)-
7a
,(
R
)-
7a
及外消旋的
7a
同时在标准条件下进行反应。(
S
)-
7a
的反应速率明显快于(
R
)-
7a
,这表明催化剂对于底物具有很强的手性识别能力。(
S
)-
7a
及(
R
)-
7a
分别被立体专一地氢化为光学纯的(
S
,
S
)-
6a
及(
R
,
R
)-
6a
,展现了强烈的底物诱导作用。这些结果也与图5 中对于
7a
的动力学拆分现象相契合。对于消旋的
7a
进行催化氢化反应后,可以 50 % 的收率及 80 % 的 ee 值得到了(
S
,
S
)-
6a
,原本消旋的原料
7a
出现了对映体过量,其 ee 值为 77 %。
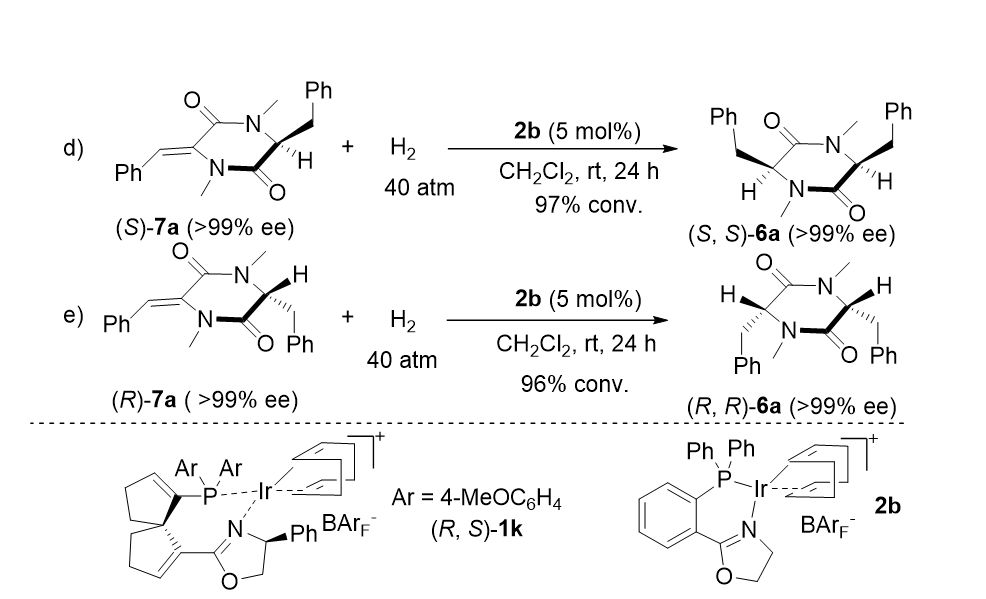
▲图7. 底物诱导现象的实验验证
为了排除手性催化剂与底物识别作用的影响,本文进一步采用非手性的 Pfaltz 型铱络合物
2b
为催化剂,进行氢化反应的尝试(图7)。光学纯的(
S
)-
7a
,(
R
)-
7a
在非手性铱络合物
2b
的催化作用下,分别以>99 % 的 ee 值得到(
S
,
S
)-
6a
及(
R
,
R
)-
6a
,并没有任何内消旋异构体的生成。由此证明
7a
氢化产物的立体构型完全由底物的立体化学控制。对于含有两个双键的标准底物
5a
的催化氢化,当中间体
7a
完全被消耗时,氢化产物
6a
的 ee 值完全由第一步氢化的对映体过量程度决定。










