现在搞科研,动不动就要接触高通量测序,想要来篇高分文章,没有高通量感觉都不太好意思,所以如果你还不知道什么是FASTQ和FASTA,那就太OUT了,其实他们在我们用的最多的PUBmed里随处可见:

FASTQ是基于文本的,保存生物序列(通常是核酸序列)和其测序质量信息的标准格式。其序列以及质量信息都是使用一个ASCII字符标示。
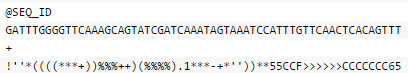
FASTQ格式中,每个序列通常有四行:第一行,序列标识以及相关的描述信息,以‘@’开头;第二行是序列;第三行以‘+’开头,后面是序列标示符、描述信息,或者什么也不加;第四行,是质量信息,和第二行的序列相对应,每一个序列都有一个质量评分,根据评分体系的不同,每个字符的含义表示的数字也不相同,比如以下:
而Fasta格式也称为Pearson格式,是一种基于文本用于表示核苷酸序列或氨基酸序列的格式。在这种格式中碱基对或氨基酸用单个字母来编码,且允许在序列前添加序列名及注释。
Fasta格式首先以大于号“>”开头,接着是序列的标识符;换行后是序列的描述信息。换行后是序列信息,文件每行的字母一般不应超过80个字符。序列中允许存在空格,换行,空行,直到下一个大于号或文件结束,表示该序列的结束。

以上两种格式蕴含了大量的生物学信息,对其游刃有余的应用对我们科研大有裨益,但很可惜,需要编程基础。今天小编给大家介绍一款神器,处理FASTA和FASTQ的工具SeqKit,window\linux系统版本都有.对于没有编程基础的小伙伴们,我们照样可以轻松操作序列文件。
该软件功能强大,小编只罗列部分模块功能,更详细功能参见软件网站:
http://bioinf.shenwei.me/seqkit/usage/。之后会讲解一些应用,请做好功课。
seqkit seq [flags] file
参数:
-p, --complement 取互补序列
--dna2rna DNA to RNA
-l, --lower-case 将序列以小写字母形式输出
-g, --remove-gaps 移除组装序列中的gap
-r, --reverse 取反向序列
--rna2dna RNA to DNA
-u, --upper-case 将序列以大写字母形式输出
-w, --line-width int 以每行指定长度输出序列 (0 for no wrap) (default 60)
举例:
seqkit seq test.fa -w 0#将此文件fasta序列转换成一行输出
seqkit seq -w 100 test.fa#将此文件fasta序列转换成100个碱基一行输出
seqkit seq --dna2rna test.fa#将此文件fasta序列dna转换成rna
seqkit seq -w 100 -p -r test.fa#将此文件fasta序列反向互补输出,每行100碱基
1、FASTQ转换成FASTA: seqkit fq2fa
举例:seqkit fq2fa reads_1.fq -o reads_1.fa
2、FASTA/FASTQ转换成tab格式。seqkit fx2tab
举例:
seqkit fx2tab test.fa>test.fa.tab.fa
seqkit fx2tab test.fq>test.fq.tab.fq
tab格式:ID sequence
1、序列碱基含量及序列长度信息统计
seqkit fx2tab [flags]
参数:
-B, --base-content value 要输出的碱基含量e.g. -B AT -B N
-g, --gc print GC content
-l, --length print sequence length
-n, --name only print names
-i, --only-id print ID instead of full head
举例:
seqkit fx2tab -l -g -n -i -H test.fa
输出结果:
#name seq qual length GC
gene1 30 40.00
2、序列长度分布统计
Usage:
seqkit stat [flags]
举例:
seqkit stat test.fa
输出结果
file format type num_seqs sum_len min_len avg_len max_len
test.fa FASTA DNA 1 30 30 30 30
seqkit grep [flags]
参数:
-n, --by-name 匹配整个序列的名字,包含description部分,而不是序列id。
-s, --by-seq 匹配序列
-d, --degenerate pattern/motif 包含简并碱基
-i, --ignore-case 忽略大小写
-v, --invert-match 输出不匹配此模式的内容
-p, 匹配模式,支持连续写多个模式,匹配任一模式即输出。如-p ^ATG -p TAA$。注意该功能仅能正向匹配,不能实现对互补链匹配。
-f, --pattern-file string 支持匹配模式写到一个文件中,如要提取的序列ID。
-R, --region string 匹配位置选择。e.g 1:12 for first 12 bases, -12:-1 for last 12 bases
-r, --use-regexp 使用正则表达式,必须加入此参数,如^匹配首端。同-p联合使用。
示例:
seqkit grep -s -r -i -p ^atg cds.fa#选取有起始密码子的序列
seqkit grep -f list test.fa > new.fa#根据ID提取序列
seqkit grep -s -d -i -p TTSAA#简并碱基使用。S 代表C or G.
seqkit grep -s -R 1:30 -i -r -p GCTGG##匹配限定到某区域
对grep的拓展,可以正反链同时匹配,输出匹配的位置。
seqkit locate [flags]
参数
-d, --degenerate pattern/motif contains degenerate base
-i, --ignore-case ignore case
-P, --only-positive-strand only search at positive strand
-p, --pattern value search pattern/motif
-f, --pattern-file string pattern/motif file (FASTA format)
举例
seqkit locate -i -d -p AUGGACUN test.fa
输出结果
seqID patternName pattern strand start end matched
cel-mir-58a AUGGACUN AUGGACUN + 81 88 AUGGACUG
ath-MIR163 AUGGACUN AUGGACUN - 122 129 AUGGACUC
六、多个序列文件比较寻找相同的序列或者ID相同的序列 seqkit common [flags]
参数:
-n, --by-name 匹配整个序列的名字,包含description部分,而不是序列id
-s, --by-seq match by sequence
-i, --ignore-case ignore case
-m, --md5 use MD5 reduce memory usage
举例:
1、By ID (default,>后面,空格之前的名字)输出ID名字相同的。
seqkit common test1.fa test2.fa -o common.fasta
2、By full name(整个序列的名字,包含description部分)。输出序列名字相同的。
seqkit common test1.fa test2.fa -n -o common.fasta
3、输出要比较的文件中序列相同的序列
seqkit common test1.fa test2.fa -s -i -o common.fasta
4、输出要比较的文件中序列相同的序列 (for large sequences)
seqkit common test1.fa test2.fa -s -i -o common.fasta --md5
如随机抽取10000条FASTQ序列做NT污染评估。同时他也可以对FASTA序列提取
seqkit sample [flags]
参数:
-n, --number int sample by number (result may not exactly match)
-p, --proportion float sample by proportion
-s, --rand-seed int rand seed for shuffle (default 11)
-2, --two-pass 2-pass mode lower memory
举例:随机抽取序列
seqkit sample -n 10000 -s 11 test1_1.fq -o sample.fq
seqkit sample -p 0.1 -s 11 test1_1.fq -o sample.fq
seqkit sort [flags]
参数:
-l, --by-length 按照序列长度排序
-n, --by-name by full name
-s, --by-seq 按照序列排序
-i, --ignore-case 按序列排序时忽略大小写
-r, --reverse 反向排序
-2, --two-pass 对于FASTA序列排序可以减少内存
举例:
seqkit sort -l test.fa
seqkit split [flags]
参数:
-i, --by-id split squences according to sequence ID
-p, --by-part int 将一个文件分割成N 份
-s, --by-size int 将一个文件按照N 条序列一个文件进行分割
-O, --out-dir string output directory (default value is infile.split)
-2, --two-pass two-pass mode to lower memory usage(only FAST)
举例:
seqkit split hairpin.fa.gz -p 4
生信人团队组建于2014年,由老祝,鑫仔,helen等组建,他们都来自同一个学校:哈尔滨医科大学,更来自同一个寝室,他们是国内最早一批专攻生物信息学专业的学生,这个时候,他们希望能将生信知识广播,让生信更有价值,生信人将自行开发免编程的可视化软件,以软件操作为实例,结合线上、线下,为广大生信小白提供一系列免费、且更实用、实操性的生信技能运用。
专题 | 生物信息学之初学者(一)
专题 | 生物信息学之初学者(二)
这几本生信入门书籍你不能不知,附下载链接











