自 Amgen--Clinical and Scientific Considerations for Biosimilars, 2018
作者 l Irene
编辑
l
Schhill
生物仿制药从2004年欧盟建立相关法规起已经有十几年的发展历史了,有人说因为可以提供有价格竞争力的病人能负担的药品所以这个市场潜力巨大,有人说因为较高的专利壁垒和比想象更多的资本投入做生物仿制药没有前途,市场众说纷纭迷雾重重。无论怎么预测城中的玩家各有各的绝活和策略,最终商场还是成败论英雄,谁能真正快速开发出能负担得起的生物仿制药并最终在市场上赢得一席之地,谁就是最终的赢家。当然在一个技术主导的行业如果没有对生物仿制药的技术和市场的深入理解,要成功也难。从这个角度来说生物制药巨头Amgen 大举进入生物仿制药市场是对自己的核心竞争力的延展,也是非常明智的策略,从它这两年在生物仿制药上的快速发展已经可见一斑了。浏览它的生物仿制药网站无意看到“Clinical and Scientific Considerations for Biosimilars”的报告, 借机和大家一起学习一下Amgen对生物仿制药的最新理解。
生物仿制药生产工艺的复杂性
与化学药物相比,生物制药的制造过程更加复杂,涉及多达200个额外的步骤。生物药物的制造过程开始于建立独特的细胞系,由细胞生产的任何药物即使生产中小的变化也会导致关键质量属性发生重大变化(如活性),生物仿制药也不例外。对于药物的认定,纯度,生物活性和稳定性而言重要的特征被称为关键质量属性。生物仿制药开发商无法获得原研产品的专有生产制造知识和细胞库,所以必须把将要开发的生物仿制药与原研参考产品进行比较对其进行定性。要开发生物仿制药,生物仿制药开发商必须从头创建生产制造工艺,包括从各种表达系统中选择构建独特的细胞系,开发特定的细胞生长培养基,生物反应器培养条件和纯化条件等。并且他们必须使用灵敏的且经过验证的分析方法对关键质量属性(例如纯度,浓度,结构和生物功能)进行分析,表征证明生物仿制药与参考产品的相似性。
成功开发生物仿制药需要在蛋白质工程,细胞系开发和大规模细胞培养方面的专业知识。
可比性和生物相似性的不同--Comparability和 Biosimilarity
|
|
产品历史
|
工艺评估
|
分析研究
|
非临床研究
|
临床研究
|
|
可比性:相同生产商的相同产品
|
修改的工艺:简略的可比
|
√
|
√
|
√
|
|
|
|
新的工艺:全面的可比
|
√
|
√
|
√
|
√
|
√
|
|
生物相似性:不同生产商的新产品
|
生物类似物工艺
|
?
|
?
|
√
|
√
|
√
|
证明所提出的生物仿制药与参考产品的生物相似性通常比在同一制造商制造变更之前和之后评估产品的可比性要复杂得多。
这是因为修改自己制造工艺的制造商对自有产品和现有工艺有广泛的知识和信息,包括已建立的控制和验收参数。相反,建议的生物仿制药的制造商可能会有不同的生物仿制药的生产制造工艺(例如不同的细胞系、原材料、设备、工艺、过程控制和验收标准),而这与直接了解原研药的工艺不一样。因此,即使在ICH Q5E中描述的一些科学原理可以应用于证明生物相似性,一般而言,建立生物相似性比建立生产变更前后的可比性需要更多的数据和信息。
全球生物制药的批准路径

生物仿制药的批准路径与化仿药不同,批准基于对来自临床前定性和临床实验整体数据包的评估。欧盟的法规路径是欧洲和其他国家生物仿制药批准的依据。拉美11个国家基于WHO的指南制定了生物仿制药的批准流程。亚洲包括日本、韩国在内的6个国家建立的生物仿制药的法规批准路径,中国于2015年发布了生物仿制药开发和评估指南。WHO 2015年开发了法规评估生物仿制药的路线图。
美国生物制品的批准流程
|
产品
|
申请类型
|
申请路径
|
要求
|
|
生物制品
|
生物制剂许可证申请
|
351(a)
|
纯度,安全性和效力的完整的临床评估
|
|
生物仿制药申请
|
351(k)
|
证明所建议的生物仿制药与351(a)批准的参考产品在安全性,纯度和效力方面高度相似(即没有临床意义上的差异)
对于可互换的生物制剂,申请的生物仿制药首先必须被批准为生物仿制药,并且由于转换(对于多次给药的产品)对任何患者具有相同的临床结果,不会对安全性或疗效产生额外风险。
|
生物仿制药的命名
目前生物仿制药的命名全球没有形成统一的标准,
2017年1月USFDA颁布的命名指南中要求对任何生物药物必须在非专有名后添加4个无任何意义的小写字母组成的后缀。
日本药监部门规定生物仿制药通用名后加”BS”加生产厂商的名字。 WHO对生物仿制药的命名没有特别的规定,目前只有对糖基化和非糖基化产品的名称命名上有区别,非糖基化的氨基酸顺序一样的生物药物享有统一的通用名,糖基化的产品通用名的根是一样的但会加上一个希腊字母来区别不同的糖基化形式。FDA最新批准了的几个生物仿制药的命名如etanercept-szzs,adalimumab-atto等等。
适应症的外推
生物仿制药厂家不需要对原研药的所有适应症进行临床试验,在美国生物仿制药厂家可以提供科学的证据对已批准的原研药品的适应症进行外推。这意味着一种临床研究的生物仿制药可用于某种肿瘤类型或肿瘤疾病状态在不需要提供临床数据的前提下可能被批准用于其他肿瘤类型或疾病。为了支持批准生物仿制药外推适应症,申办者需要证明生物仿制药在临床试验和推断的适应症中具有相同的作用机制、靶标结合特性、PK和生物分布,并解决任何预期的毒性或有效性的差异。
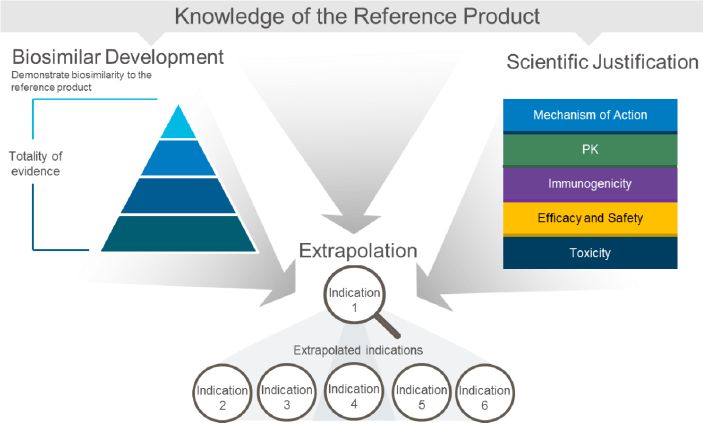
FDA的生物相似性和可替换性-Biosimilarity和Interchangeability

根据BPCIA,相对参考产品生物仿制药可被批准为“生物仿制药”或“可互换生物制剂”。要获得“可互换性“的称号,产品必须证明其可以产生与任何给定患者的参考产品相同的临床结果。2017年1月,美国FDA发布了指南草案,指出哪些数据足以认定生物制品可以与参比产品互换。
证明可互换性所需的数据类型和数量由美国FDA根据具体情况逐一确定, 这取决于多种因素的组合,例如生物制品的复杂性,其物理化学特性,参考产品的临床经验以及免疫原性风险的潜在可能性。
欧盟的生物仿制药批准经验—药物认定上的细微差异与临床意义的差异
欧盟2004年建立了生物仿制药相关法规,2005年开发了生物仿制药的批准路径, 2006年批准了第一个生物仿制药,目前欧盟已经批准了9类生物仿制药。大部分生物仿制药的申请获得了欧盟上市的许可,当然一些申请在欧盟在审评中提出了担忧后被拒绝或撤销。
在一个例子中,EMA基于仿制药与参比产品治疗组之间存在显著统计学生物物理差异和临床变异(PK,功效和耐受性)拒绝α-干扰素生物仿制药的批准申请。CHMP担心的其他问题包括:杂质,稳定性数据不足,不良事件率中的显著差异,及在免疫应答测试和制造工艺中缺乏足够的验证。同样,在产品未能证明与参考产品的PD相似之后,欧盟的三个人类胰岛素生物仿制药候选申请被撤销。在一项将生物仿制药与参考产品进行比较的预授权临床研究中,与接受参比产品的患者相比,接受生物仿制药的更多患者产生了非中和抗GH抗体。接下来,改变生物仿制药产品的制造工艺的纯化步骤,免疫原性问题得到了解决。
非常重要的一点是法规部门并没有期望参比药物与生物仿制药完全一致。
例如,就PTM(翻译后修饰)如糖基化,磷酸化,乙酰化和唾液酸化而言,欧盟生物仿制药与参考产品方面存在差异。这些生物仿制药与参比制剂之间的生物物理变化在临床上并没有观察到统计学上的显着影响。 2013年,EMA批准了第一种生物仿制药抗肿瘤坏死因子(TNF)mAb,尽管在体外测定中检测到生物活性的一些差异,但是
这种差异因为不影响在与实验模型更相关的生物仿制药中的活性病人的病理生理状况,而未被解释为具有临床意义和影响。
小结
难而不知,知而不难,要做好生物仿制药需要深厚的蛋白质工程,细胞系开发和大规模细胞培养方面的专业知识,同时还要与时俱进。期待生物仿制药的下一个高峰的到来。
参考
https://www.amgenbiosimilars.com/pdfs/Clinical_and_Scientific_Considerations_for_Biosimilars_USA-BIO-058953.pdf
说明:本文所有内容包括图片来自参考链接,仅供学习,如有不妥请及时联系小编,我们会妥善处理,如需引用请说明原出处,特此感谢。
欢迎加入小编团队成为小编一员
请加小编微信号:wuwenjun7237
如有技术解读、行业洞见愿意分享
欢迎投稿到小编邮箱:
[email protected]











