(一)现场核查发现缺陷的总体情况
经对313个药品注册申请的现场核查报告进行分析,共发现5111条缺陷项。其中临床部分4583条,平均每个临床试验机构发现问题6条;生物分析部分528条,平均每个生物样本分析单位发现问题4.4条。依据《药物临床试验数据现场核查要点》对缺陷进行分类,发现缺陷条款数量最多的部分依次为:临床试验过程记录及临床检查、化验等数据溯源方面(占28.1%)、方案违背方面(占12.0%)、试验用药品管理过程与记录方面(占11.6%)和安全性记录、报告方面(占10.1%),共发现缺陷3161项,占61.8%(见图1)。
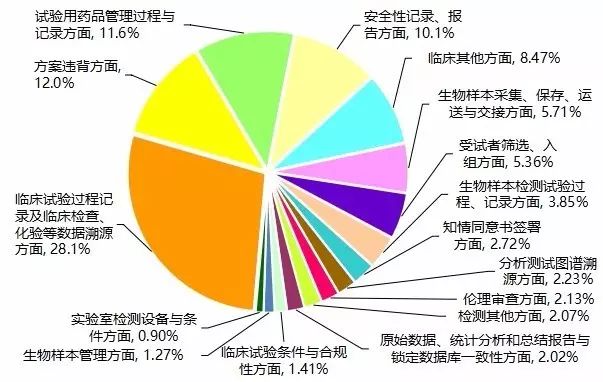
图1. 核查缺陷分布情况
(二)高频次缺陷条款分布情况
对临床部分现场核查发现的4583项缺陷汇总分析,频次最高的缺陷条款见图2。
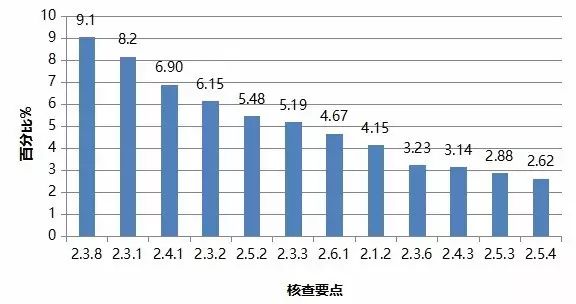
图2. 高频次缺陷条款分布情况(临床部分)
对分析测试部分现场核查发现的528项缺陷汇总分析,频次最高的缺陷条款见图3。
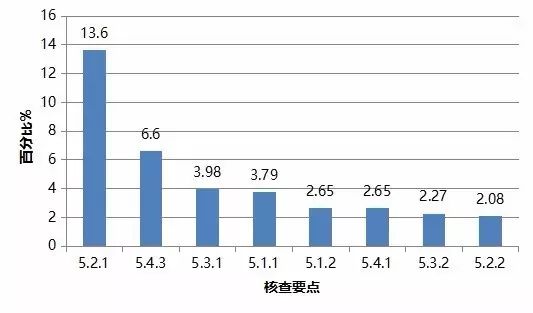
图3. 高频次缺陷条款分布情况(分析测试部分)
(三)发现主要问题
1. 临床试验过程记录及临床检查、化验等数据溯源方面
☆ 受试者体检单上性别栏显示为男性,但X线片图像显示为女性;报告单总的报告时间和人员信息被裁掉;脉搏治疗前后一致,不合常理;研究病历显示受试者已死亡,仍继续有访视记录和检验报告;多例受试者试验的血生化化验单中的部分数据(主要为肝肾功能指标)与医院LIS系统中的数据存在严重差异;
☆ 临床试验仅有病例报告表,无原始病历记录;
☆ 数据不可溯源,受试者血常规、尿常规等数据无法溯源;
☆ 不同受试者入出组的血生化报告值完全相同;
☆ 心电图图谱、报告真实性存疑,同一受试者试验前后波形不一致;
☆ 主要疗效指标、次要疗效指标无法溯源等等。
☆ 中心实验室样品放置时间超过样品在该条件下的稳定时间;样品运输过程中无温控记录;检测报告单上无检测人和复合人的签名;样品报告时间与样品接收间隔数天;样品被连续转包导致检测周期过长等等。
2. 方案违背方面
☆ 修改入组检查结果,使其符合方案规定入组标准;
☆ 漏记方案规定禁用药物、合并用药;
☆ 给药方式、给药剂量及采血时间偏离试验方案;
☆ 未按方案随访至规定日期等等。
3. 试验用药品管理过程与记录方面
☆ 试验用药品不真实,参比制剂的包装规格和药片外观与试验制剂完全一致;
☆ 直接将试验制剂用作参比制剂,造成两者等效的假象;
☆ 药品发放和回收数量不一致;
☆ 药品管理员未被授权;
☆ 药品运输过程中的温度超出规定温度;
☆ 无药品回收记录;
☆ 药品接收和发放记录单无药品批号或批号不一致;
☆ 药品发放记录单中修改不规范等等。
4. 安全性记录、报告方面
☆ 发生AE,CRF和总结报告中未录入;
☆ 修改AE与试验药物相关性的判断;
☆ 未按规定上报SAE;
☆ AE漏记等等。
5. 生物样本检测试验过程、记录方面
☆ 仪器还未购买就有了检测数据;缺乏生物样本预处理、保存、转运以及 LC-MS/MS 、离心机使等关键部分的记录;
☆ 样本分析过程记录原始记录缺失,相关记录为事后整理补充填写,分析过程记录等是后期整理得到,没有原始分析记录;
☆ 隐瞒弃用试验数据,原始记录本中未记录;
☆ 图谱文件与血样没有关联性,两者之间的联系可随意更改且无法追溯;
☆ 部分报告数据与原始图谱计算所得数据不一致;
☆ 存在不合理手动积分等等。
6. 分析测试图谱溯源方面
☆ 方法学验证及生物样本测试的稽查轨迹中均多处出现分析测试系统日期反复更改、重复检测后用同一文件名命名并覆盖原有图谱;
☆ 从质谱图谱中发现选择性使用符合要求的标准曲线和质控样品,并在 Audit trail 中发现将被采用的样本的序号(Sample ID)修改成连续序列;
☆ 多个时间点样品编号与对应的图谱中的文件名编码从小到大的顺序颠倒;
☆ 部分受试者分析序列采集过程有中断,质控样本重复进样,部分样本重分析而没有随行质控,但数据被采用;
☆ 多个样本测试数据文件的采集时间有重叠;
☆ 仪器被转卖,分析测试图谱无法溯源;
☆ 选择性使用数据;
☆ 分析测试图谱无法溯源等等。
7. 伦理委员会主要问题
☆ 伦理委员会未对上报的SAE进行审查;
☆ 试验方案修改未经伦理委员会审查;
☆ 项目接收组长单位伦理批件,而未见任何相关的记录;
☆ 未见针对该项目伦理委员会会议审查的原始记录;
☆ 伦理委员会会议审查记录原始记录本内容与整理后的会议纪要内容不符。










