第
1
章 序言
本指导原则是对已批准的口服固体制剂有效成分、疗效、用法
/
用量及剂型均完全相同、仅主成分含量不同的仿制制剂,在进行生物等效性试验时的相关规定。主要为保证不同含量的制剂,等量服用时的生物等效性。
根据已批准制剂的处方变更级别,实施不同的试验。
第
2
章 专业用语
基准处方
已通过临床试验验证了其有效性与安全性的制剂处方;或是已通过人体试验验证了与原研制剂生物等效的仿制制剂处方。
参比制剂
取
3
批已批准制剂,在下列溶出介质①或②中进行试验,采用第
4
章项下的溶出试验参数(仅做桨板法、
50
转,测定
6
个以上单位样品),选取中间那条溶出曲线的批次作为参比制剂。但是,对于
A
级的处方变更,如果参比制剂的质量标准或试验方法中已拟定了溶出试验,也可按照该试验条件进行试验。介质①或②中,
3
批次均在
15
分钟内平均溶出
85%
以上的情况,任一批次均可确定为参比制剂。
①
质量标准或试验方法中已拟订溶出度检验项目,则选取该溶出介质。
②
在第
4
章项下的溶出试验介质中,至少有
1
个批次样品平均溶出率达
85
%以上,则选取溶出速率最慢的溶出介质;如果任何
1
个批次样品,在所有溶出介质中平均溶出率均未达
85
%,则选取溶出速率最快的溶出介质。
试验制剂
与参比制剂含量不同的制剂,最好是今后工业化生产规模条件下生产出来的,或不少于今后工业化生产规模的
1/10
。同时,今后工业化生产的制剂工艺必须与用于体外溶出试验同等性判定的样品的生产工艺相一致,以保证两者具有相同的内在品质和生物利用度。
缓控释制剂,应与参比制剂在大小、形状、密度、释放机理等特性上没有显著性差别,且必须与参比制剂的溶出行为相似。关于溶出行为相似性的判定,在
1997
年
12
月
22
日医药审发第
487
号文《仿制药生物等效性试验指导原则》以及
2012
年
2
月
29
日医药审发
0229
第
10
号《仿制药生物等效性试验指导原则(
2012
版)》的第
3
章,
B. IV. 4.
中已有详细阐述和规定。
难溶性药物制剂
遵循仿制药指导原则第
3
章,
A. V. 3. 3)
项下规定。
第
3
章制剂处方变更级别与所需验证试验
1
.制剂处方变更级别
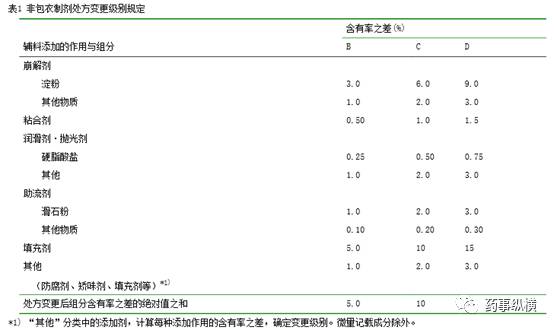
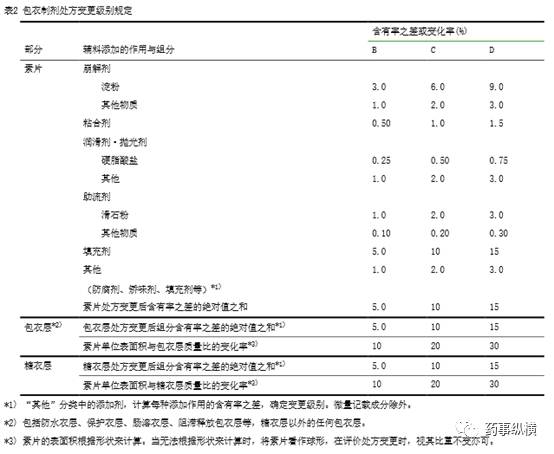
处方变更级别是以基准处方为基准计算出来的。根据表
1
或表
2
“辅料添加作用与组分表”检索出相应级别。
B
级以下的算作
B
级、大于
B
级小于
C
级的算作
C
级、大于
C
级小于
D
级的算作
D
级、大于
D
级的算作
E
级。
除治疗窗狭窄的制剂、缓释制剂、肠溶制剂外,如处方变更为如下①
-
③项,则变更级别与表
1
、表
2
无关,执行
A
级变更。
①
除微量成分外,所有成分的组成比均相同的变更【注】;
②
主成分含有率之差在
0.5%
以内变更时,为保证制剂质量不变而增减填充剂含量的变更;
③
“其他”分类中的成分,以同样添加作用进行
1.0%
(含有率之差的绝对值之和)以内的变更(例:矫味剂种类变更)。
除治疗窗狭窄的药物外,包衣层质量占素片质量
7.0%
以下的包衣层变更,遵照附录
3
,包衣层对溶出没有影响的情况下,则与表
2
的包衣层变更级别无关,执行
B
级变更。
制剂的处方变更水准以最大变更为准。对于肠溶制剂,如果具有肠溶机能的基本单位,其直径从不到
4mm
变更到
4mm
以上,或是从
4mm
以上变更到
4mm
以下,一律执行
E
级。追加《仿制药生物等效性试验指导原则》第
3
章,
B.
Ⅱ
.1.
的食后给药试验,根据《仿制药生物等效性试验指导原则》第
3
章
A.
Ⅱ
.2.
进行判断。
【注】对于包衣制剂,不但要求薄膜层,糖衣层的所有成分的组成比均相同,还要求素片单位表面积与包衣层的质量比(或与糖衣层的质量比)相同。
2
.所需验证试验
在实施
BE
试验时,应尽可能地给予一致的剂量,原则上不能超过用药上限。当给药量无法一致时,将测得的药物动力学参数按照给药剂量予以相应地校正亦可。(但仅限于给药量与药物动力学参数呈线性关系的制剂品种。)
溶出试验时,
1
个溶出杯中的投药量,原则上不能超过试验制剂与参比制剂中含量高的制剂的含量。
A
级
参比制剂质量标准或试验方法中拟定有溶出度检查的,选用该试验条件进行溶出试验(至少测试
12
单位制剂);参比制剂质量标准或试验方法中没有拟定溶出度检查的,遵循第
4
章所述的原则进行溶出试验,根据第
5
章所列标准判定溶出曲线为同等的,则可将两者看作“生物等效”。
如溶出试验结果显示两者不等效,则需遵循《仿制药生物等效性试验指导原则》进行生物等效性试验研究。
B
级
按照第
4
章所述的原则进行溶出试验。其中,包衣层对溶出试验没有影响的,如果包衣层处方变更,且参比制剂在本《指导原则》所规定的任一溶出试验条件下,平均溶出率不及
85%
时,可以进行
A
级规定的试验。根据第
5
章所列标准判定溶出曲线为同等的,则可将试验制剂与参比制剂看作生物等效。
如溶出试验结果显示两者不等效,则需遵循《仿制药生物等效性试验指导原则》进行生物等效性试验研究。
C
级
普通制剂与肠溶制剂
按照第
4
章所述的原则进行溶出试验(难溶性药物除外)。根据第
5
章所列标准判定为同等的,则可将两者看作生物等效。但表
3
中所示药物制剂(表中为治疗窗狭窄药物),不仅要求在第
4
章中任何一个溶出试验条件下,试验制剂与参比制剂均要在
30
分钟内平均溶出率达
85%
以上,且还要根据第
5
章中所列标准判定溶出曲线为同等的,才能将两者看作生物等效。如溶出试验结果显示两者不等效,则需遵循《仿制药生物等效性试验指导原则》进行生物等效性试验研究。
难溶性药物制剂
遵循《仿制药生物等效性试验指导原则》进行生物等效性试验研究。
缓控释制剂
按照第
4
章所述的原则进行溶出试验研究(治疗窗狭窄药物除外)。根据第
5
章所列标准判定溶出曲线为同等的,则将两者看作生物等效。如溶出试验结果显示两者不等效,则需遵循《仿制药生物等效性试验指导原则》进行生物等效性试验研究。
治疗窗狭窄药物制剂
遵循《仿制药生物等效性试验指导原则》进行生物等效性试验。
D
级
普通制剂
按照第
4
章所述的原则进行溶出试验(难溶性药物制剂和治疗窗狭窄药物制剂除外)。第
4
章中任何一个溶出试验条件下,试验制剂与参比制剂均能在
30
分钟内平均溶出率达
85%
以上,且根据第
5
章中所列标准判定溶出曲线为同等的,才能将两者看作生物等效。如溶出试验结果显示两者不等效,则需遵循《仿制药生物等效性试验指导原则》进行生物等效性试验。
难溶性药物制剂和治疗窗狭窄药物制剂
遵循《仿制药生物等效性试验指导原则》进行生物等效性试验。
肠溶制剂和缓控释制剂
遵循《仿制药生物等效性试验指导原则》进行生物等效性试验。
E
级
遵循《仿制药生物等效性试验指导原则》进行生物等效性试验。

第
4
章 溶出试验
按照《仿制药生物等效性试验指导原则》中第
3
章
A
、
V
以及第
3
章
B. IV
项下进行试验。但浆法的转数,不能用
75
转代替
50
转。如果在水中药物会吸附在添加剂或溶出杯上,可以用
0.2%
的氯化钠溶液替代水。对于需添加吐温
-80
的难溶性药物,其添加浓度应在
0.1%
以下。如为肠溶制剂,需追加以下溶出试验。使用十二烷基硫酸钠时,药物的溶解度不能高于吐温
-80
最大浓度时的溶解度。
试验:用
0.01mol/L
磷酸二氢钠溶液和
0.005mol/L
枸橼酸溶液调制成
pH6.0
的溶出介质,体积
900ml
,桨板法,
50
转。
第
5
章 溶出行为同等性判定
对于各溶出试验条件、当满足于下列
(1)
和
(2)
中所列标准时,即可判定两者溶出行为同等。但在规定时间内,至少一个试验条件下,普通制剂与肠溶制剂应达到参比制剂平均溶出率的
85%
;缓控释制剂应达到参比制剂平均溶出率的
80%
。
“规定时间”是指《仿制药生物等效性试验指导原则》中第
3
章
A
、
V. 2
或第
3
章
B. IV. 2
项下规定的试验时间。采用
f2
因子评价时,比较时间点的确定参照附件
1(2)
。对于普通制剂与肠溶制剂,如果参比制剂或试验制剂中任何一个出现溶出滞后现象时,可以采用延迟时间对溶出曲线予以校正(附件
2
),再进行延迟时间后溶出曲线的比较。如果校正溶出曲线,应保证试验制剂与参比制剂的平均延迟时间差在
10
分钟以内。
比较时间点未达到
15
分钟的情况,可以认为比较时间为
15
分钟,进行溶出行为的评价。如果需要进行滞后时间校正,比较时间点
15
分钟被认为是校正前时间。
肠溶制剂,在
pH1.2
的溶出介质中,只需评价既定试验时间(
2
小时)内的溶出率既可。
(
1
)
平均溶出率
①
参比制剂在
15
分钟以内平均溶出率达
85%
以上时
试验制剂在
15
分钟以内平均溶出率也达
85%
以上;或
15
分钟时,试验制剂与参比制剂两者的平均溶出率差在±
10%
以内。
②
参比制剂在
15
~
30
分钟平均溶出率达
85%
以上时
对应参比制剂平均溶出率
60
%和
85
%附近两个时间点,试验制剂与参比制剂两者的平均溶出率差在±
10%
范围内;或是
f2
因子大于
50
。
③
参比制剂在
30
分钟内平均溶出率达不到
85%
时
只要满足下列任何一个条件,仍可被判定溶出曲线具有同等性。
普通制剂与肠溶制剂
a.
参比制剂的平均溶出率在规定时间内达
85%
以上时,对应参比制剂平均溶出率
40
%和
85
%附近两个时间点,试验制剂与参比制剂两者的平均溶出率差在±
10%
以内;或
f2
因子大于
50
。
b.
参比制剂平均溶出率在规定时间内达
50%
以上但未达
85%
时,对应最终时间点和参比制剂在最终时间点平均溶出率的
1/2
所对应的时间点,试验制剂与参比制剂两者平均溶出率的差在±
8%
以内;或
f2
因子大于
55
。
c.
参比制剂平均溶出率在规定时间内达不到
50%
时,对应于最终时间点和参比制剂在最终时间点平均溶出率的
1/2
所对应的时间点,两者平均溶出率的差在±
6%
以内;或
f2
因子大于
61
。
但是,规定试验时间内,参比制剂的平均溶出率低于
10%
时,则在规定试验时间内,两者平均溶出率差在±
6%
范围内。
缓控释制剂
a.
参比制剂在规定时间内平均溶出率达
80%
以上时,对应参比制剂平均溶出率
30
%、
50
%和
80
%附近的三个时间点,两者平均溶出率的差在±
10%
以内;或是
f2
因子大于
50
。
b.
参比制剂平均溶出率在规定时间内达
50%
以上但未达到
80%
时,对应最终时间点和参比制剂在最终时间点平均溶出率的
1/2
所对应的时间点,两者平均溶出率的差均在±
8%
以内;或是
f2
因子大于
55
。
c.
参比制剂平均溶出率在规定时间内达不到
50%
时,对应最终时间点和参比制剂在最终时间点平均溶出率的
1/2
所对应的时间点,两者平均溶出率的差在±
6%
以内;或是
f2
因子大于
61
。
但是,规定试验时间内,参比制剂的平均溶出率低于
10%
时,则在规定试验时间内,两者平均溶出率差在±
6%
范围内。
(
2
)
对于个体溶出行为的规定
在最终比较时间点,试验制剂的个体溶出率应满足以下任何一个标准。










