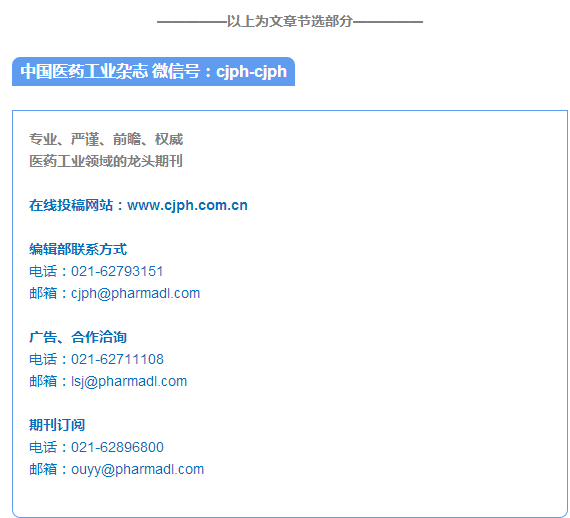
不对称氢化反应在手性药物合成中的应用
Applications of Asymmetric Hydrogenation in the Synthesis of Chiral Drugs
陈才友1,董秀琴1*,张绪穆1,2*
(1. 武汉大学化学与分子科学学院,湖北武汉 430072;2. 南方科技大学化学系,广东深圳 518055)
摘要:
不对称催化氢化反应在过去几十年中得到了空前的发展,目前发展的种类多样的高效手性膦配体大大拓展了不对
称氢化反应的底物适用范围。基于均相不对称氢化反应的高原子经济性、高反应活性、反应条件温和以及对环境绿色友好等优点,如今,均相不对称氢化反应被广泛地应用于手性药物的合成中。本文简要介绍了不对称氢化反应中一些代表性的手性膦配体,同时介绍了应用高效高选择性不对称氢化反应合成手性药物的代表性实例。
关键词:
不对称氢化;手性膦配体;铑;钌;铱;催化剂;药物合成

不对称氢化反应是指在氢化催化剂和氢气的
条件下,将潜手性的化合物,如烯烃、酮或亚胺等转化为相应的手性还原产物的反应。由于不对称氢化反应使用绿色的氢气作为还原剂,因此相较于其他不对称催化方法,不对称氢化反应是用于构建手性分子最为高效和高原子经济性的方法之一[1]。近年来,工业和人类日常生活中对手性药物、香料、农用品等日益增长的需求,极大地促进了不对称氢化反应的发展[2]。基于不对称氢化的重要作用,在不对称氢化领域做出卓越贡献的著名科学家Knowles 和Noyiri 被授予了2001 年的诺贝尔化学奖。
根据催化剂的不同,不对称氢化反应可以分为
两大类,即均相催化氢化和异相催化氢化。相对于均相催化氢化反应,异相催化氢化反应有更为久远的历史[3]。早在1956 年,Akahori 等就报道了Pd催化的氮杂内酯类化合物的异相催化氢化反应[4]。基于这一开创性的工作,后来也发展了经过负载的催化剂(Raney Ni[5] 和Pt[6]) 催化的酮类化合物的不对称氢化。虽然异相催化剂可以取得中等或者优异的对映选择性,但是其反应活性一般较差,这一缺点限制了异相催化剂在不对称氢化反应中的实际应用[3]。随后人们发展了更为高效的均相催化剂,很好地克服了异相催化剂活性低的缺点。均相催化剂在不对称氢化反应中表现出了优异的对映选择性和反应活性[7],因而,均相催化氢化在过去的50
年中得到了空前的发展,也成为了现代有机合成化
学的经典方法学之一[8]。
最早的均相不对称氢化反应是Knowles 和
Horner 等在20 世纪60 年代末期发展的[9]。通过将Wilkinson 催化剂[RhCl(PPh3) 3] 中的三苯膦配体用具有光学活性的手性膦配体甲基正丙基苯基膦取代,Knowles 和Horner 等发展出了一种手性的Rh均相催化剂,随后他们考察了这一手性Rh 络合物在潜手性烯烃的不对称氢化反应中的应用[9]。虽然ee 值较差( 最高只有15% ),但他们第一次实现了均相不对称氢化反应,而这一开创性的研究也极大地促进了均相不对称氢化反应的发展。
随后,Dang 等于1971 年发展了一种具有C2
对称性的螯合手性双膦配体DIOP( 图1) [10]。手性DIOP 配体在α- 脱氢氨基酸及烯酰胺类底物的不对称氢化反应中取得了较好的对映选择性( ee 值最高达88% ),这也是第一例高效、高选择性的手性双膦配体[ 11]。DIOP 配体的发现促进了具有C2 对称性的手性双膦配体的快速发展。最具代表性的例子是Knowles 等于1975 年发展的手性DIPAMP双膦配体( 图1),在α- 脱氢氨基酸的不对称氢化反应中表现出了优异的对映选择性( ee 值高达96% )[12]。
自Dang(DIOP 配体) 和Knowles(DIPAMP 配
体) 等的工作以来,很多优异的手性膦配体被发展并成功地应用于不对称氢化反应中。在此期间,Halpern 等[13] 和Brown 等[14] 也对Rh 催化的不对称氢化反应的机理进行了细致的研究,这也进一步促进了均相不对称氢化反应的发展。然而,20 世纪80 年代以前的研究工作大多集中于Rh 催化剂,研究的底物类型也局限在α- 脱氢氨基酸及其衍生物[1]。1980 年,Miyashita 等发展出了另一个里程碑式的配体BINAP( 图1)[15]。BINAP 配体的发展,极大地拓宽了不对称氢化反应的底物范围。同时,Noyori 等发展了高效的Ru-BINAP 催化剂。这一催化剂的发现,使得酮类化合物的不对称氢化反应得到了长足的发展。
1993 年,Matt、Sprinz 和Dawson 等发展了
一类新型的手性N,P- 配体(Phox 配体,图1) [ 16]。Phox 类型的配体在Ir 催化的不对称氢化反应中得到了广泛的应用。与传统配体不同,该类配体可适用于非官能团化烯烃的高效、高选择性不对称氢化反应[17]。这一发现进一步拓宽了不对称氢化反应的底物范围。
如今,成百上千的手性膦配体被成功地发展出
来,这些配体在学术研究和工业上均得到了广泛的应用。大量配体的发展使得不对称氢化反应的金属不仅仅限于Rh、Ru 或者Ir,其他金属如Pd、Pt、Ti、Zr 有时也可以作为有效的金属前体[1]。同时,不对称氢化的底物范围也大大拓展,不仅α- 脱氢氨基酸及其衍生物、官能团化烯烃、酮类化合物,甚至一些非官能团化底物的不对称氢化反应都可以很好地实现。均相不对称氢化反应的快速发展为手性药物的合成提供了极大的便利。基于均相不对称氢化反应的高原子经济性、高反应活性、反应条件温和以及对环境绿色友好等优点,如今均相不对称氢化反应被越来越多地应用于手性药物的合成中。本文主要概述在不对称氢化领域中的一些代表性手性膦配体,并简要介绍不对称氢化反应在手性药物合成中的一些代表性应用。
1 不对称氢化反应配体发展概述
1.1 早期DIOP 和DIPAMP 手性膦配体
1.2 BINAP 配体及其Ru 络合物
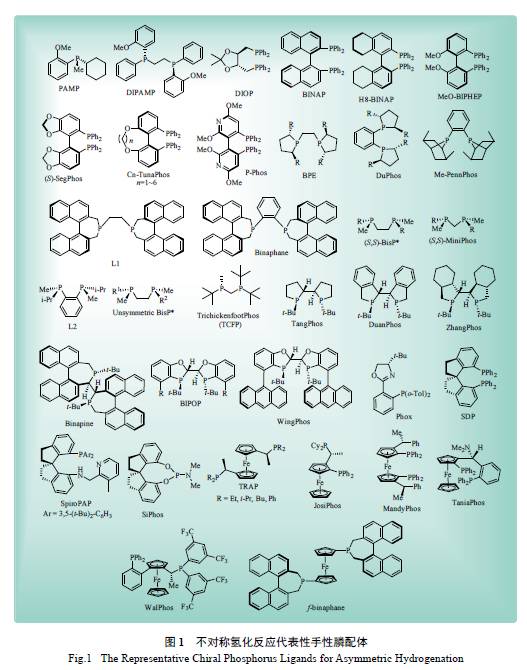
Miyashita 等发展了另一个里程碑式的配体,即
轴手性联芳基双齿膦配体BINAP[ 15]。最初,该配体被应用在Rh 催化的α- 脱氢氨基酸及其衍生物的不对称氢化反应中,展现出了优异的对映选择性(ee值高达>99% ) [19]。然而,Rh-BINAP 催化剂在这类底物的不对称氢化反应中的反应活性并不高,并且该催化剂的底物适用范围并不广。真正使BINAP配体得到广泛应用的原因是Ru-BINAP 络合物的发现。1986 年,Noyori 和Takaya 等发展了经典的Ru-BINAP 催化剂( 图2),该催化剂在一系列官能团化烯烃的不对称氢化反应中均表现出了优异的反应活性及对映选择性[20]。
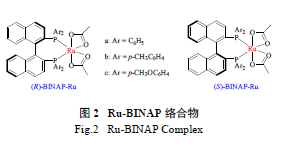
20 世纪90 年代中期,关于BINAP 配体的另一
大突破性研究进展是Ru-BINAP/ 双胺类络合物的发现[21]。Ohkuma 等发现,Ru/BINAP-Dpen 和Ru/BINAP/Daipen 络合物( 图3) 催化体系在简单酮类化合物的不对称氢化反应中具有超高的反应活性和对映选择性。当Ru/BINAP/Daipen 络合物中配体上P 原子的取代基为3,5- 二甲基苯基时,该络合物在一系列酮类化合物的不对称氢化反应中均展现出超高的反应活性及对映选择性。在苯乙酮的不对称氢化反应中,Ru/BINAP/Daipen 催化剂的最高转化数(TON) 超过了1 000 000,最高转化频率(TOF) 超过了600/s[22]。此外,该催化剂应用广泛[23],其特征为:①具有高反应活性及对映选择性;②具有广泛的底物适用范围;③可以选择性氢化羰基而保留C=C 键。
1.3 轴手性联芳基双齿手性膦配体
在BINAP 配体的研究基础上,其他研究小组
也相继发展了多种高效的轴手性联芳基双膦配体( 图1)。例如,本课题组发展了H8-BINAP 配体。在Ru 催化的不饱和羧酸的不对称氢化反应中,该配体展现了更好的对映选择性[24]。Schmid 等发展了MeO-BIPHEP 配体[25],并成功用于许多Ru 催化的不对称氢化反应中。研究表明,双芳基骨架之间的二面角对氢化结果有很大影响。Takasago 公司
发展了一类比BINAP 配体的二面角更小的轴手性
双膦配体SegPhos,在Ru 或Rh 催化的不对称氢化反应中应用广泛,在一些底物的不对称氢化反应中,该配体甚至展现出比BINAP 配体更好的对映选择性[26]。
1.4 BPE 及DuPhos 手性双齿膦配体
20 世纪90 年代早期,Burk 等发展了一类新
型的具有广泛底物适用性的手性膦配体BPE 及DuPhos( 图1) [ 30],在一系列底物的不对称氢化反应中均展现出优异的反应活性及对映选择性。Rh-BPE 或Rh-DuPhos 络合物在烯酰胺、α- 脱氢氨基酸酯、β- 羰基酯、烯醇乙酸酯、α,β- 不饱和羧酸及衣康酸类底物中均展现出优异的对映选择性[31]。
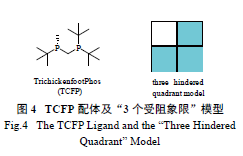
1.5 基于3 个受阻象限模型发展的TCFP 配体
自从Kagan 等发展C2 对称性的手性双膦配
体DIOP 以来,C2 对称性的配体设计理念已被广泛应用,然而,研究发现该设计理念并不是至关重要的。一些非C2 对称性的配体在不对称氢化反应中也展现了很好的对映选择性。2004 年,
Hoge 等发展了一种新型的非C2 对称性的双膦配体
TrichickenfootPhos (TCFP)[42],TCFP 引入了“3 个受阻象限”模型(three hindered quadrant model,图
4) 的设计原则。TCFP 配体中的3 个位阻很大的叔
丁基使TCFP-Rh 络合物中的3 个象限非常拥挤,而具有较小位阻的甲基则使另一个象限比较空旷。TCFP 配体这一独特的设计原则使TCFP-Rh 催化剂在一些特殊底物的不对称氢化反应中也有着非常好的对映选择性。
1.6 TangPhos 类型手性双齿膦配体
基于BisP* 配体在不对称氢化反应中的发
展, 本课题组发展了另一种里程碑式的配体TangPhos( 图1)[43]。为了增加t-Bu-BisP* 配体的刚性,本课题组引入了膦杂五元环结构。同时,在金属/TangPhos 络合物中,TangPhos 配体的2 个叔丁基有效地屏蔽了络合物中的2 个象限,而另外2 个象限则很开阔。这种完美的C2 对称性及位阻效应使得该配体在不对称氢化反应中得到了广泛的应用,在一系列底物的不对称氢化反应中均展现出非常优异的对映选择性[44]。
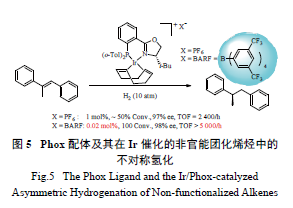
1.7 Phox 配体
虽然手性Rh 及Ru 催化剂在不对称氢化反应
中应用广泛,但是该类催化剂的底物范围一般局限于官能团化的烯烃,即与烯烃C=C 键相邻的位置上要有一个导向基团,而不适用于没有导向基团的非官能团化烯烃的不对称氢化反应,因而这些非官能团化烯烃的不对称氢化反应具有很大的挑战性。Lightfoot 等发展了一类高效的Phox 配体( 图1),完美地解决了这一挑战性的难题[50]。初步研究表明,[Ir(Phox)(COD)]+PF6- 络合物在非官能团化的烯烃(E)-1,2- 二苯基丙烯的不对称氢化反应中可获得非常好的对映选择性( ee 值高达97% );但是其催化活性并不高,这主要是由不对称氢化过程中催化剂的失活导致的。随后的研究表明,配阴离子对Ir-Phox 络合物的催化活性影响非常大。将PF6- 阴离子换成非极性、弱配位、大位阻的BARF阴离子后,Ir-Phox 络合物的催化活性大大提高。例如,在(E) -1,2- 二苯基丙烯的不对称氢化反应中,催化剂的最低用量仅0.02 mol% ( 图5)[51]。
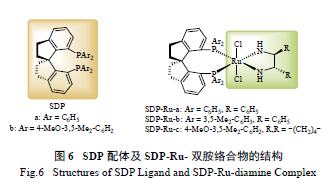
1.8 螺手性膦配体
2003 年,Xie 等发展了一类基于螺二氢茚骨架
的螺手性双膦配体SDP( 图6) [52]。螺二氢茚骨架具有C2 对称性,同时刚性强、易于修饰。该配体骨架的发现促进了螺手性膦配体的快速发展。研究表明,螺手性双膦配体SDP 在Ru 催化的芳香酮的不对称氢化反应中有着很高的反应活性及对映选择性。SDP-Ru- 双胺络合物在苯乙酮的不对称氢化反应中TON最高达100 000,ee 值高达99% ( 图6)[52]。
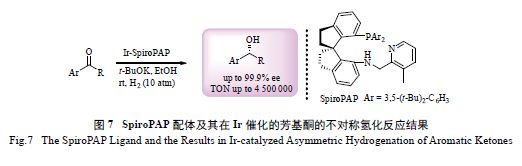
周其林等此前发展的螺手性-P,N- 配体SiPhox,
在不对称氢化反应中与金属Ir 生成的催化物易生成二聚体或三聚体,从而导致失活,因此Ir-SiPhox络合物的反应活性不是很高[53]。为了解决这一问题,2011 年,该课题组又发展了新型P,N,N- 螺手性配体SpiroPAP( 图7) [54]。该配体有一个额外的可配位的吡啶基团,吡啶基团的存在使Ir-SprioPAP络合物非常稳定( 具有良好的空气稳定性),同时,还避免了该络合物的二聚或三聚,从而大大提高了其催化活性。Ir-SprioPAP 络合物在一系列简单酮类化合物的不对称氢化反应中应用广泛,并且展现出了非常好的反应活性和对映选择性。例如,在苯乙酮类底物的不对称氢化反应中,ee 值最高可达99.9%;此外,反应的TON 最高可达到惊人的
4 500 000[54a]。这一TON 也是目前均相催化反应中
的最高值。
1.9 基于二茂铁骨架的手性膦配体
二茂铁骨架具有高度富电子、易于修饰的特性,
此外,基于该骨架的手性膦配体具有良好的空气稳定性。基于这些优势,研究人员发展了许多基于二茂铁骨架的手性膦配体( 图1)。20 世纪90 年代,Sawamura 等发展了一类基于二茂铁骨架的手性膦配体TRAP[57],其在不对称氢化反应中的底物适用范围广泛。
2 不对称氢化反应在手性药物合成中的应用
不对称催化氢化反应在过去几十年中得到了空
前的发展。多种高效、高选择性的手性膦配体的发展大大拓展了不对称氢化反应的底物适用范围。如今,Rh、Ru 或Ir 金属催化的 α- 脱氢氨基酸及其衍生物、官能团化烯烃、酮类化合物,甚至一些非官能团化底物的不对称氢化反应都可以很好地实现。目前越来越多的不对称催化氢化反应也实现了工业化应用。如表1 所示,截至2008 年,适用于工业化应用( 工业生产、中试或实验规模) 的9 种不对称催化反应中,有6 种与不对称催化氢化反应相关[ 63]。此外,这6 种催化反应的总数也远远大于其他催化反应的总数。从表1 的数据可以看出,不对称催化氢化反应在工业化应用中已经占据了举足轻重的地位,而此类反应很大程度上与手性药物及其中间体的制备相关。本文将介绍一些不对称催化氢化反应在手性药物合成中的代表性实例。
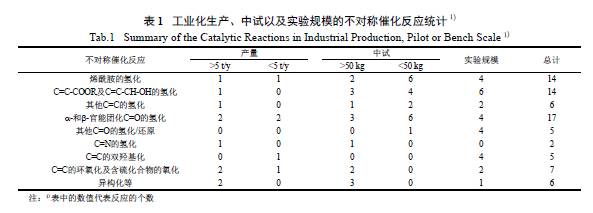
2.1 左旋多巴(levodopa,L-DOPA) 的合成
手性药物L-DOPA 是神经递质多巴胺的前体,
用于帕金森病的药物治疗。Knowles 等于1975 年发展并运用手性DIPAMP 双膦配体后, 实现了L-DOPA 的不对称合成[64]。如图8 所示,其关键步骤为Rh/DIPAMP 络合物催化的α- 脱氢氨基酸的不对称氢化反应,在催化剂当量S/C>10 000 的条件下,ee 值高达96%。所得的氢化产物经过脱保护即可制得L-DOPA。这也是第一例将不对称催化反应应用于工业化生产的实例。
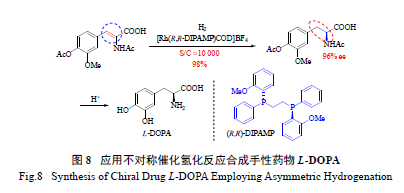
2.2 香茅醇及薄荷醇的工业合成
1986 年发现的经典的Ru-BINAP 催化剂在C=C
键的不对称氢化反应中展现出了优异的反应活性及对映选择性[15,20],已成功用于香茅醇(citronellol)的合成( 图9)。不同构型的香叶醇(geraniol) 及其异构体橙花醇(nerol) 在(S)-BINAP-Ru 催化下,可分别氢化得(R) 或( S) - 构型的香茅醇;而在(R)-BINAP-Ru 催化下,则分别得到(S) 或(R)- 构型的香茅醇[65]。值得注意的是,该氢化反应的活性很高,催化剂当量很低(S/C=50 000)。此外,反应的对映选择性和化学选择性也很好,ee 值最高达99%,同时只有邻近羟基的C=C 键发生了选择性氢化,远离羟基的C=C 键则得到了很好的保留。
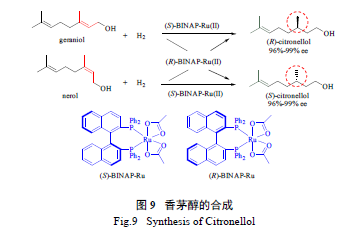
BINAP 配体除了可应用于香茅醇的合成,在工
业上的一个更大用途是合成L- 薄荷醇(L-menthol)( 左旋薄荷醇具有薄荷香气并有清凉的作用,通常可用作食品添加剂;而在医药上薄荷醇常有清凉止痒作用)。如图10 所示,其关键步骤为,经由Rh/BINAP 络合物催化的烯烃不对称异构化反应制备手性香茅醛(citronellal) 的烯胺中间体,该中间体再经后续转化即可制得L- 薄荷醇[66]。目前,工业上通过该方法合成的L- 薄荷醇的产量每年已经超2 000 吨。
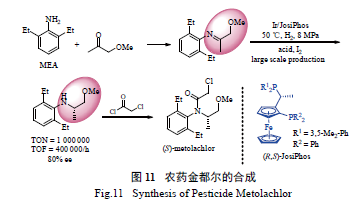
2.3 农药金都尔( 异丙甲草胺,metolachlor) 的合成
1994 年报道的JosiPhos 配体已被大规模地应
用于工业化生产中,最大规模的用途是合成手性农药( S) - 金都尔[67]。如图11 所示,其关键步骤为Ir/JosiPhos 络合物催化的亚胺的不对称氢化反应。该反应使用了催化量的I2 作为活化剂,具有超高的反应活性,TON 最高可达1 000 000,TOF 最高可达40 000/h。
2.4 萘普生(naproxen) 和布洛芬(ibuprofen) 及其
衍生物的合成
手性药物萘普生和布洛芬及其衍生物具有抗
炎、解热、镇痛作用,传统方法一般采用拆分法,原料利用率较低。目前已发展了多种适用于α- 取代丙烯酸不对称氢化反应制备萘普生和布洛芬的高效催化体系,其中具有代表性的如图12 所示。
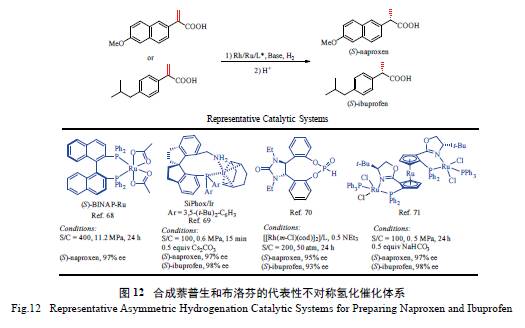
由图12 可知,虽然上述催化体系能以较好的
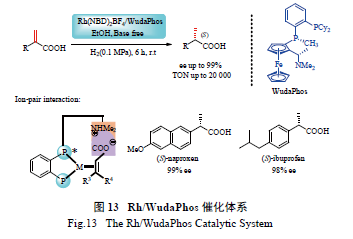
对映选择性及催化活性制得萘普生和布洛芬,但是反应大多需加入当量的碱,同时反应压力较高。最近,本课题组通过引入离子键相互作用,发展了一类新型的基于二茂铁的手性双膦配体WudaPhos( 图13)[72],相应的Rh/Wudaphos 催化体系在α- 取代丙烯酸的不对称氢化反应中展现了超高的反应活性及对映选择性(ee 99%,TON 20 000)。如图13 所示,该催化体系中,配体的二甲胺基可以与α- 取代丙烯酸的羧基形成离子键相互作用,从而大大增加了催化体系的反应活性及对映选择性,同时反应条件温和( 室温、常压),不需加碱作添加剂。该催化体系可分别以高达99%及98%的ee 值制得萘普生和布洛芬。
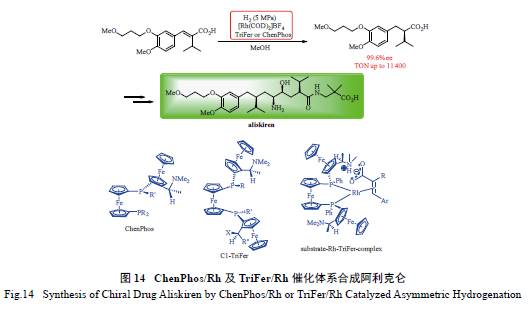
2.5 阿利克仑(aliskiren) 的合成
手性药物阿利克仑是第二代肾素抑制剂,能
降低肾素活性,起到降血压和治疗心血管疾病的作用。最近,通过引入离子键相互作用,陈卫平和McCormack 等发展了高效的ChenPhos[73] 及TriFer[74] 配体( 图14),ChenPhos/Rh 及TriFer/Rh催化体系在α- 异丙基-β- 芳基丙烯酸的不对称氢化反应中展现了优异的反应活性及对映选择性。该催化体系中,配体的二甲胺基可与底物的羧基形成离子键相互作用,从而不需额外添加碱,且反应条件温和。ChenPhos/Rh 及TriFer/Rh 催化体系能以高达99.6%的ee 值及高达11 400 的TON 制得用于合成
阿利克仑的重要中间体,再经该中间体即可方便地
合成阿利克仑。
2.6 普瑞巴林(pregabalin) 的合成
手性药物普瑞巴林可用于治疗神经疼痛及癫
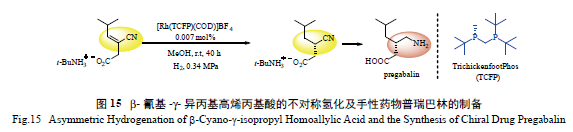
痫,2008 年全球销售额已超25 亿美元。目前为止,已经发展了多种有效的合成工艺用以制备普瑞巴林,其中包括拆分、去对称化后霍夫曼降解及不对称氢化合成法[75]。这3 种合成方法中,不对称氢化法最为简洁高效。2004 年,前文介绍的Hoge 等发展的TCFP-Rh 络合物在β- 氰基-γ- 异丙基高烯丙基酸的不对称氢化反应中展现了非常高的反应活性及高达99%的ee 值,相应的氢化产物经过一步转化即可制得普瑞巴林( 图15)[41]。
2.7 它拉那班(taranabant) 的合成
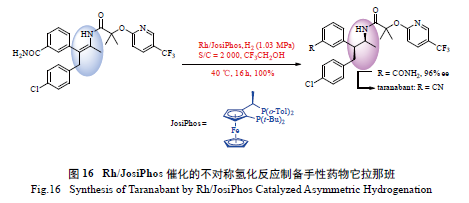
手性药物它拉那班是由Merck 公司开发的用于
治疗肥胖症的药物,它的一种高效合成路线可采用不对称氢化反应作为关键步骤。如图16 所示,其关键步骤为Rh 催化的四取代烯胺的不对称氢化反应。JosiPhos 配体在该底物的不对称氢化反应中展现了优异的反应活性及对映选择性,相应的含有2个手性中心的产物的ee 值高达96% ( 再经一步重结晶操作,ee 值可高达99.5% ),该手性产物经与
三聚氰氯反应等即可制得它拉那班[76]。
2.8 抗菌药碳青霉烯(carbapenems) 的合成
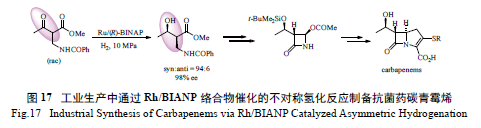
碳青霉烯类抗菌药具有广泛的抗菌活性,是治
疗严重细菌感染的最主要抗菌药之一。如图17 所示,日本Takasago 公司运用Rh/BINAP 络合物催化的不对称氢化反应,实现了该药物分子的大规模工业生产。其关键步骤为Rh/BINAP 络合物催化的β- 羰基酯类化合物的动态动力学拆分不对称氢化反应[77]。该反应可将消旋的β- 羰基酯类化合物转化为含有2 个手性中心的单一构型产物,非对映选择性和对映选择性均非常好。相应的氢化产物经过转化即可制得氮杂环丁酮关键中间体,从而实现碳青霉烯类药物分子的合成。后续研究发现,在该反应中,
Rh/SegPhos 络合物可表现出更好的非对映选择性和
对映选择性[78]。
2.9 盐酸西那卡塞(cinacalcet hydrochloride) 的合成
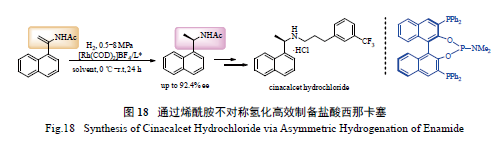
手性药物盐酸西那卡塞可用于治疗慢性肾病患
者的继发性甲状旁腺功能亢进症,也可用于治疗甲状旁腺癌患者的高钙血症。2006 年,本课题组发展了一类基于轴手性联苯骨架的三齿手性膦配体( 图18),该配体在邻位取代α- 芳基烯酰胺的不对称氢化反应中展现出优异的反应活性及对映选择性,值得注意的是,该配体在Rh 催化的α-1- 萘- 烯酰胺的不对称氢化反应中的ee 值高达92.4% [ 79]。经过该氢化产物可方便地合成盐酸西那卡塞。
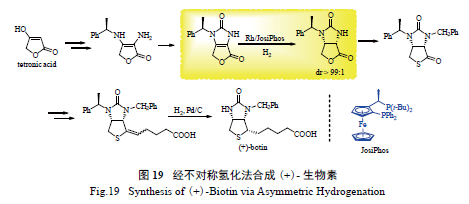
2.10 (+)- 生物素[(+)-biotin] 的合成
(+) - 生物素是水溶性生物素B 的一种,是酶
催化羰基转移反应中的助剂,是活细胞中必不可少的生长因子,对人类和动物健康起着重要作用。龙莎(Lonza) 公司通过Rh 催化不对称氢化法,实现了其高效合成[80]。如图19 所示,该路线从价廉的季酮酸出发,经后续转化,可方便地得到氢化前体。关键步骤为不对称氢化反应,该反应中Rh/JosiPhos络合物表现出了优异的选择性, 产物的dr 值>
99 ∶ 1,龙莎公司也成功将该氢化反应实现了吨级
规模。从相应的氢化产物出发,即可制得(+) - 生物素。
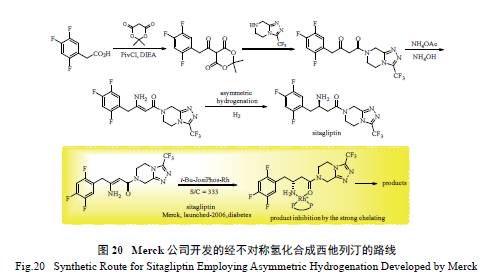
2.11 西他列汀(sitagliptin) 的合成
手性药物西他列汀是美国Merck 公司开发的
二肽基肽酶- Ⅳ抑制剂,临床用于治疗2 型糖尿病。如图20 所示,Merck 公司开发了通过不对称氢化反应合成西他列汀的有效路线,其关键步骤为Rh/t-Bu-JosiPhos 络合物催化的不保护烯胺的不对称氢化反应[81]。该路线虽然能有效地合成西他列汀,但在不对称氢化步骤中,Rh 催化剂的用量较高,S/C 仅为333,成本昂贵。必须使用高催化当量的
Rh 催化剂的原因是,生成的氢化产物对Rh 催化剂
存在螯合作用,从而抑制了催化剂的活性( 图20)。
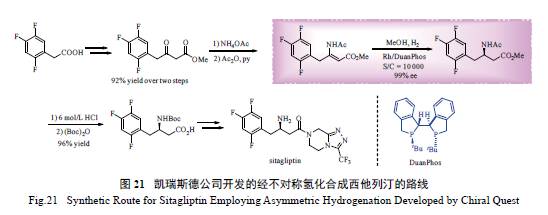
为了克服这一缺陷,凯瑞斯德公司开发了一
条更为高效的、利用不对称氢化反应作为关键步骤的合成路线,如图21 所示。由于不保护的烯胺中间体氢化后对催化剂存在毒化作用,该公司选用乙酰基保护的β- 脱氢氨基酸酯为底物,从而有效地避免了氢化产物对催化剂的毒化。采用这一策略,Rh/DuanPhos 络合物在该不对称氢化反应中展现了优异的反应活性及对映选择性,催化剂用量大大降低,S/C 高达10 000,同时ee 值高达99% [82]。目前,该关键中间体( 氢化产物) 已实现了几十吨规模的工业合成。
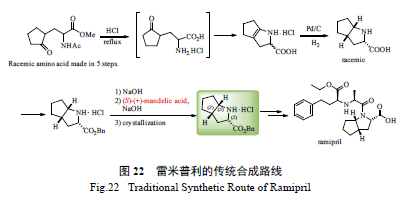
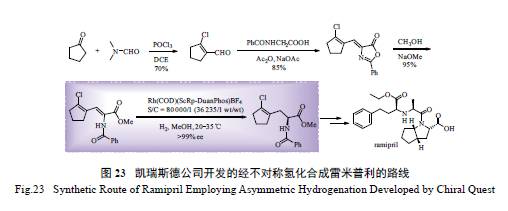
2.12 雷米普利(ramipril) 的合成
雷米普利由德国Hoechst 公司开发,1989 年在
法国首次上市,临床用于治疗高血压和心力衰竭。传统合成路线如图22 所示,其关键中间体的合成需10 步反应,且需后期拆分,产率最高只有40% [83]。该法合成效率低,会产生大量废料,污染严重。凯瑞斯德公司开发了更为高效的新合成路线,如图23 所示。反应中,关键的醛中间体通过Vilsmeier-Haack 反应高效制备而得,然后再经Erlenmeyer 反应便可高收率制得用于氢化的中间体。这一新合成路线的关键步骤为Rh/DuanPhos 催化的苯甲酰基保护的α- 脱氢氨基酸酯的不对称氢化反应,反应的催化当量低至S/C 80 000,同时产物的ee 值高达99%以上[84],从而有效地避免了传统的低效拆分方法。目前,该关键氢化产物中间体的产量已超过
几十吨。
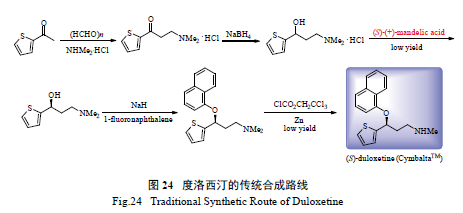
2.13 度洛西汀(duloxetine) 的合成
盐酸度洛西汀是Eli Lilly 公司开发的去甲肾上
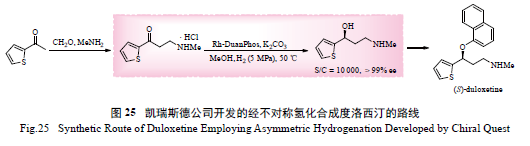
腺素再摄取抑制剂,临床用于治疗抑郁症。其传统合成方法如图24 所示,共需5 步反应,关键手性中间体需通过拆分获得,最后一步需去甲基,反应条件苛刻,同时容易混入杂质,因而合成效率较低[85]。凯瑞斯德公司发展了如图25 所示的全新合成路线,其关键中间体通过Rh/DuanPhos 催化的不对称氢化反应制得,催化剂当量低至S/C 10 000,同时ee 值高达99%以上[45b],该路线只需3 步反应,避免了效率较低的拆分和条件苛刻的脱甲基步骤,简单高效。目前,凯瑞斯德公司已实现了度洛西汀几十吨量的合成。
3 结语
不对称催化氢化反应在过去几十年中得到了空
前发展,目前已经发展了很多高效、高选择性的手性膦配体,这些手性膦配体的发展大大拓展了不对称氢化反应的底物适用范围。如今,Rh、Ru 或Ir金属催化的 α- 脱氢氨基酸及其衍生物、官能团化烯烃、酮类化合物,甚至一些非官能团化底物的不对称氢化反应都可以很好地实现。基于均相不对称氢化反应的高原子经济性、高反应活性、反应条件温和以及对环境绿色友好等优点,如今,均相不对称氢化反应被广泛地应用于手性药物的合成。相对于传统的合成方法,采用不对称氢化反应的新方法
合成手性药物通常更为直接高效,避免了传统拆分
方法的低效步骤,从而大大降低了成本。不对称氢化这一高效的有机合成方法也将被越来越多地应用于手性药物的合成中。
作者简介:陈才友(1989—),男,博士研究生,专业方向:不对称
氢化、氢甲酰化。
Tel:13164615180
E-mail:[email protected]
通信联系人:张绪穆(1961—),男,教授,博士生导师,主要从事
不对称氢化、氢甲酰化合成方法的研究。
Tel:13927474971
E-mail:[email protected]
董秀琴(1985—),女,博士,副研究员,主要从事新
型、高效的手性膦配体的研究开发及不对称氢化和氢甲酰化合成方法的
研究。
E-mail:[email protected]