
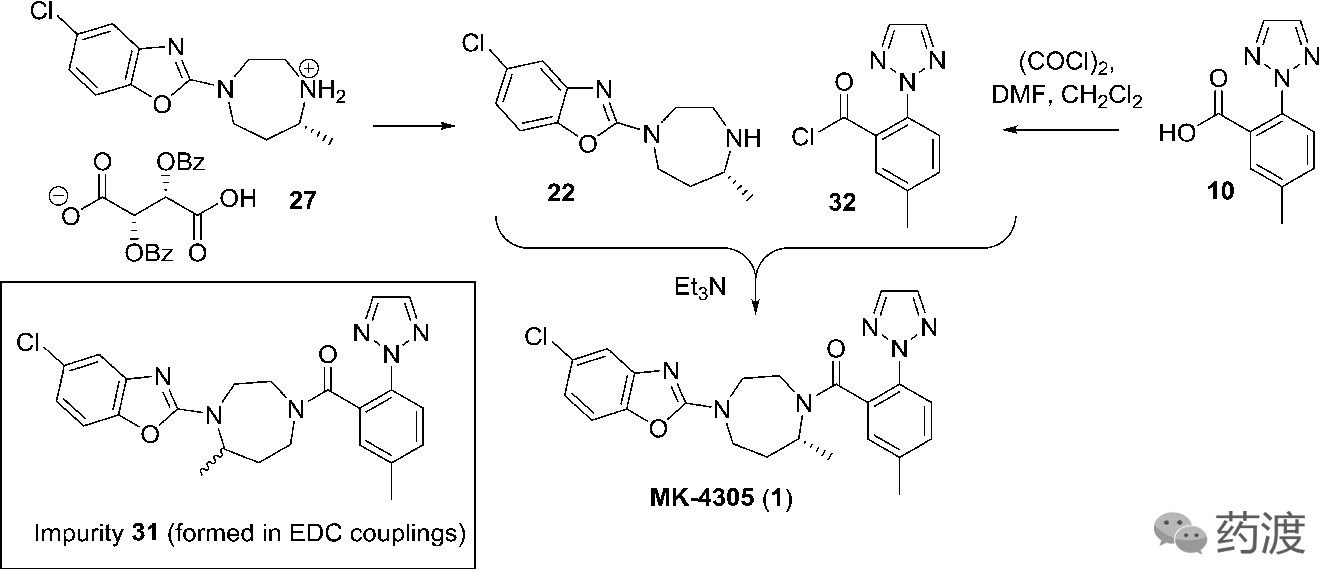
失眠是任何年龄个体均较为常见的一类睡眠问题。最常见的是在入睡、睡眠维持和/或晨间早醒方面的问题。失眠不仅对个人的社交、职业、学业等功能造成显著损害,还可诱发或加重精神及身体疾病,例如,失眠、疼痛和抑郁常形成恶性循环。
鉴于目前在失眠病因病理、可行的生物标记物以及最佳疗法的确定等方面的局限,治疗失眠症(insomnia)的新药研发主要由国外医药公司主导,国内相应的新药研发经验有限,所以本文较为详细的介绍了Suvorexant的研发历程,从先导物的发现到工艺路线的优化再到最终生产工艺的确定。
Suvorexant是由Merck公司研发,于2014年在美国上市,用于治疗失眠症。作为食欲素(Orexin)受体拮抗剂,Suvorexant是该类药物中首款获得批准的药物。
食欲素(Orexin)由Yanagisawa在1998年发现。它是由下丘脑分泌的一种饥饿调控信号,因其强烈的促食欲作用而得名。食欲素A (Orexin A)和食欲素B (Orexin B)是一种神经肽类物质,均作用于G蛋白偶联受体食欲素受体OX1R和食欲素受体OX2R。其中,OX1R与食欲素A的结合能力强于食欲素B,而OX2R与食欲素A和食欲素B的结合能力相当。随后就有研究发现患有发作性嗜睡病的犬类是由于其表达为OX2R的基因发生了突变,进而导致OX2R的功能紊乱。这一研究表明食欲素(Orexin)不但影响机体的摄食行为,还参与了睡眠-觉醒周期的调节。自此,研究者对失眠症(insomnia)的病因有了新的认识,即失眠症(insomnia)还可能是由于不恰当觉醒造成的。
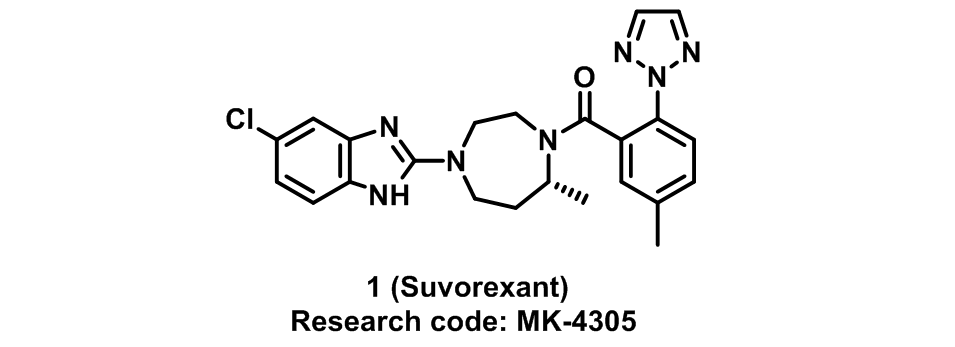
考虑到食欲素(Orexin)在睡眠-觉醒周期中的作用,Merck公司开展了对食欲素受体拮抗剂的研究。经过一系列化合物的筛选,发现化合物
2
对两种食欲素受体有较好的活性。考虑到化合物
2
的部分理化性质较差(如低溶解度和高clogP值)和较高的肝代谢率,研究人员在化合物
2
的分子骨架上做了进一步的优化,得到化合物
3
。化合物
3
在保持较好的活性同时,适当的降低了其clogP值,但不妨碍其透过血脑屏障。在对化合物
3
的进一步研究中发现,
3
在大鼠和犬中有很高的清除率和很低的口服利用度。与此同时,研究人员发现化合物
4
没有这些问题。但对其进行代谢研究发现,化合物
4
的一种代谢产物具有很强的亲电性,倾向于与谷胱甘肽(GSH)结合。将化合物
4
中氟代喹唑啉结构替换成氯代苯并噁唑结构,得到化合物
1
,即现在的
Suvorexant
,能够有效的解决代谢产物问题,同时又能保持良好的药学活性、理化性质、口服利用度和药代动力学。
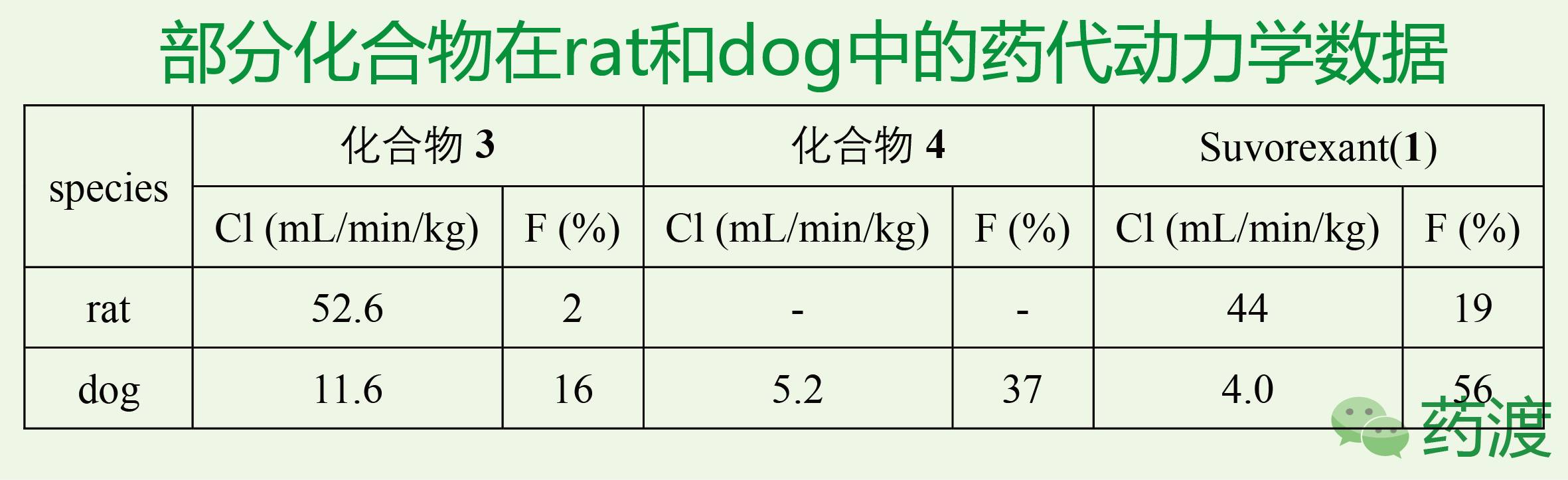
Suvorexant的早期合成路线(route1)
最初,Merck的药物化学家通过单Boc保护的乙二胺5与甲基乙烯基酮反应,随即原位CbzCl保护得到化合物
7
。选择性的脱去Boc后,通过还原胺化后,再次Boc保护得到diazepane
8
。由于得到的化合物
8
是消旋体,研究人员拿到R-和S-的对映体进行单独的研究发现,R构型的
8
对食欲素受体(OXR)有更好的药学活性。所以通过手性制备液相分得R构型的
8
,三氟乙酸脱去Boc,然后与
10
生成酰胺
13
。氢化条件下脱去Cbz,与化合物
15
发生取代反应,得到Suvorexant (
1
)。化合物
10
则是通过2-碘-4-甲基苯甲酸
11
与三唑在CuI和微波条件下得到,但是这个反应的区域选择性不好(以55:45的比例得到
10:12
),幸好可以通过色谱对两者进行分离。
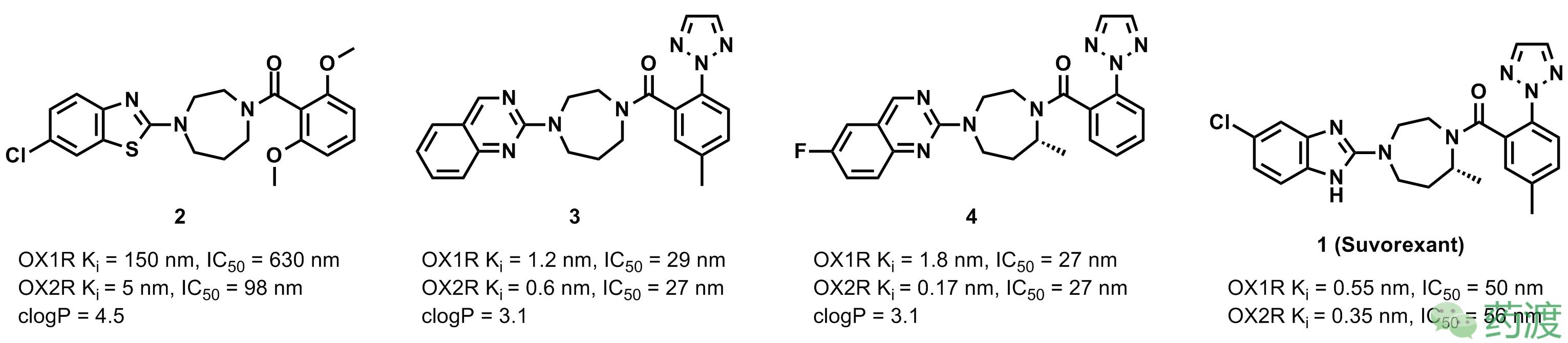
基于Suvorexant (1)良好的类药性,研究人员对Suvorexant (1)进行了动物试验。数据表明,Suvorexant (1)对大鼠、犬和非人的灵长类动物有很好的促睡眠作用,并且有很强的剂量依赖性。在临床前的毒理试验中,Suvorexant (1)也表现很好。所以Suvorexant (1)很快的被推进临床阶段。
随着试验的推进,需要大量的高质量的Suvorexant (1)样品,早先的合成路线暴露出诸多缺陷。所以研究人员对route1进行评估,提出了以下几个需要改进的地方。总结如下:
①整个合成路线中使用了大量的保护基团,增加了不必要的步骤;
②对消旋体
8
使用液相分离,效率很低;
③第一步反应中,产物
6
也会和底物甲基乙烯基酮发生aza - Michael加成,产生杂质;
④化合物
10
的制备反应中,区域选择性很差,需要通过色谱进行分离;
⑤最终分离得到的API的性状不好,过滤困难,熔点偏低。
针对以上所提出的问题,研究人员开始对路线进行重新设计,以期能更易进行放大。
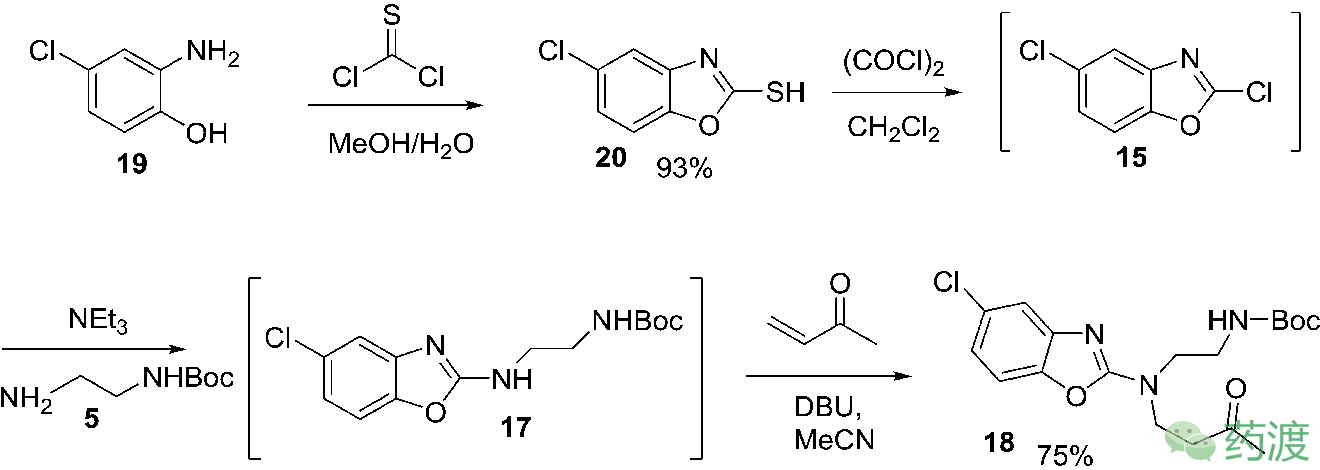
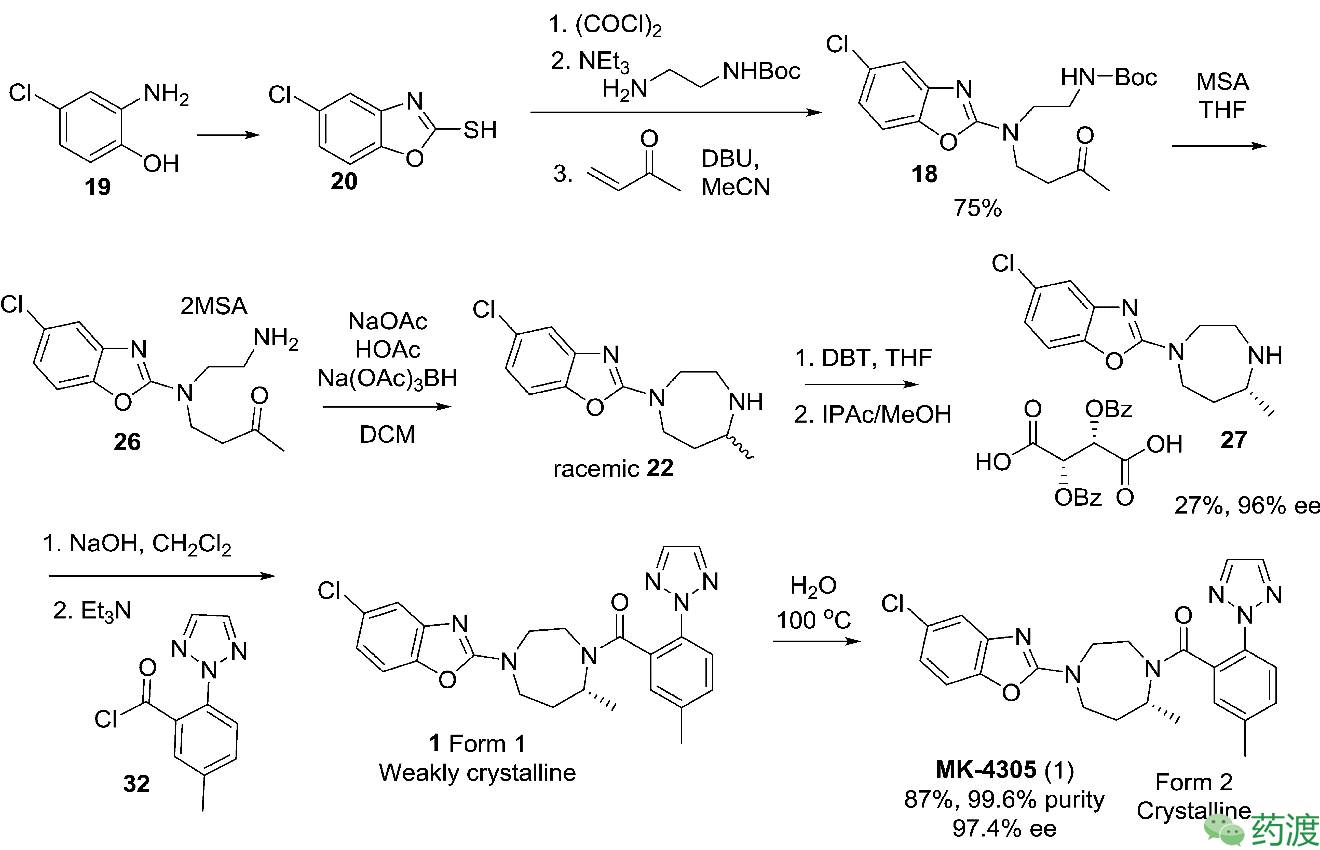
1.氯代苯并噁唑结构的早期引入
首先针对第一步反应进行设计,考虑将氯代苯并噁唑结构在路线的早期引入,具体如下图所示:
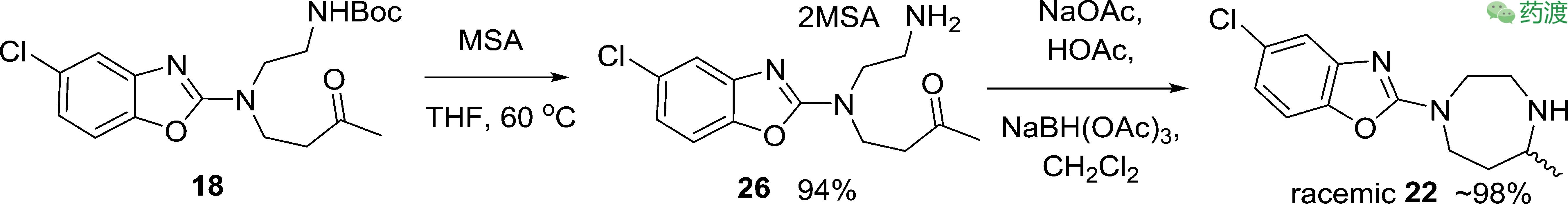
2.还原胺化和手性拆分
顺利拿到化合物
18
后,准备进行分子内的还原胺化,得到消旋体
22
。
考虑到工艺放大的问题,研究人员对消旋体
22
进行了手性拆分。在对常见的手性酸和溶剂筛选后,发现使用L-二苯甲酰酒石酸与消旋的二胺
22
在THF中能以76%ee分得所需立体构型的
22
。随后,针对这一体系做了进一步的研究。研究人员先拿到手性纯的二胺
22
,并制备相应的L-二苯甲酰酒石酸盐
2
7
和
28
,测定
27
和
28
在一些常见溶剂中的溶解度,以期能对两者达到更好的分离效果。
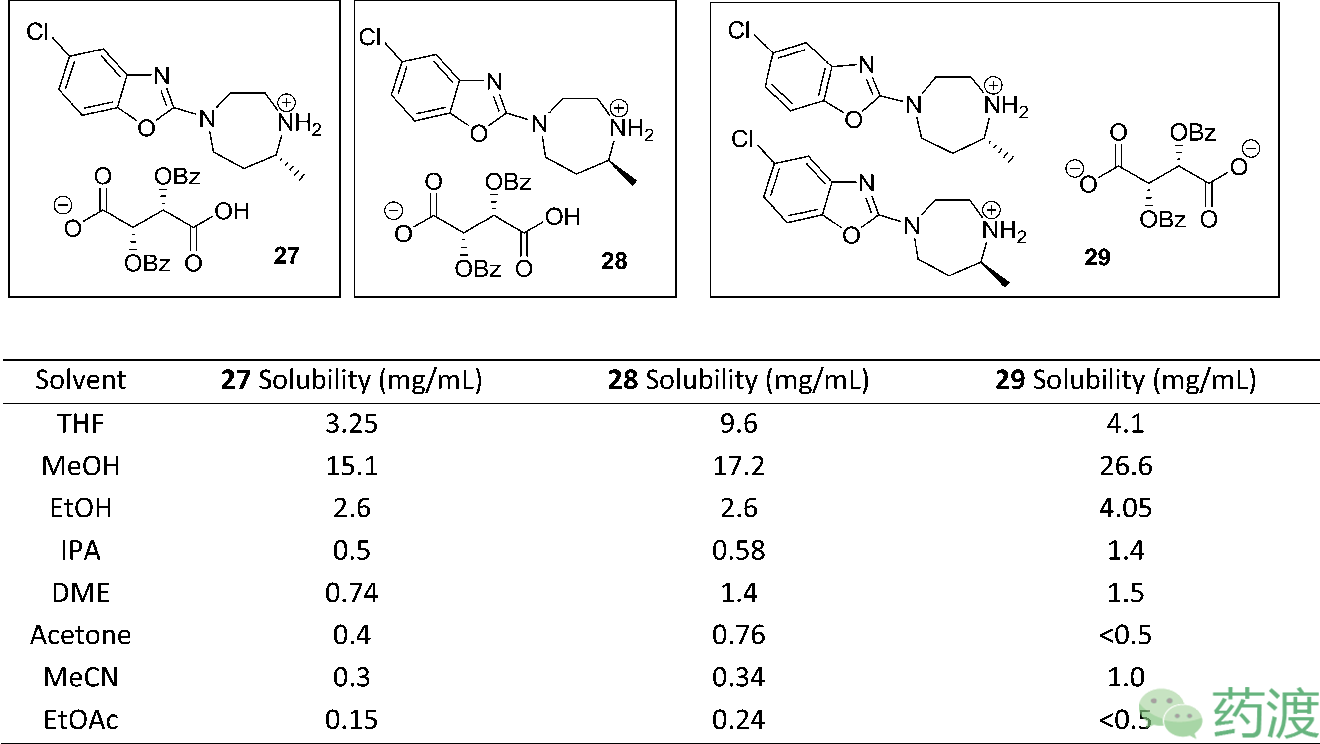
在研究中发现,二胺
22
与L-二苯甲酰酒石酸能以2:1的比例形成盐
29
。而
29
在THF中的溶解度与
27
的相当,这就是只能以76% ee分得所需立体构型的原因所在。在后续实验中,将消旋的二胺
22
缓慢的滴加到1.5 eq的L-二苯甲酰酒石酸体系中,能以38%的收率,74% ee分得所需立体构型,并且可以放大到公斤级。在拿到中等ee值的盐后,需要对它进行纯化,以提升光学纯度。最终确定在醋酸异丙酯和甲醇的混合溶剂体系中打浆,能将光学纯度提升到96%。
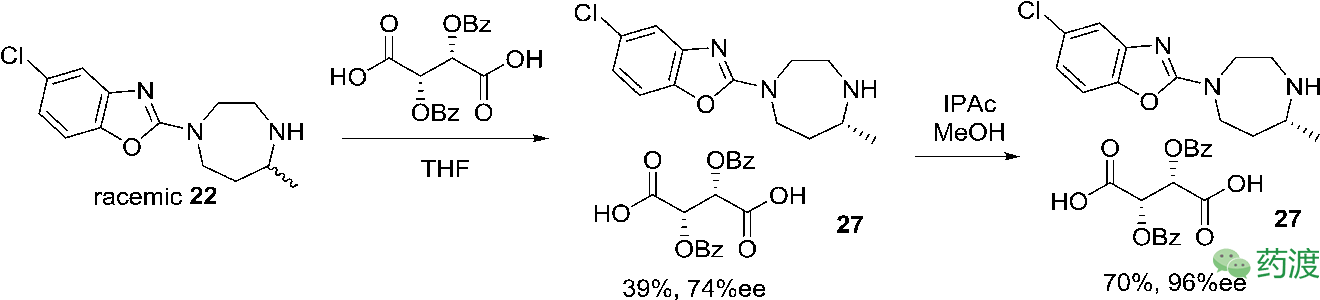
3.酰胺的形成和晶型研究
将L-二苯甲酰酒石酸盐
27
转化成相应的胺
22
后,再与酰氯
32
反应,能以88%的收率,99.5%的纯度,96.4% ee得到API。
在route 1中,制得的API物理性状不好,过滤困难。考虑到晶型对API的重要性,研究人员对晶型做了进一步的研究。Merck的研究人员将早期制备得到的晶型命名为晶型 I,他们发现将API(晶型 I)与水在近100℃件下打浆,能够得到晶型II。晶型II的性质明显优于晶型I,更适合用于后期的放大开发。但在做两者间的稳定性研究发现,晶型II于室温条件下在大多数溶剂中打浆都会转化成晶型 I,水是个例外。所以,在中试放大时,将晶型I分离后,于100
℃
条件下在水中打浆,降温后能以97%的收率,99.6%的化学纯度和97.4%的ee值分得晶型II。

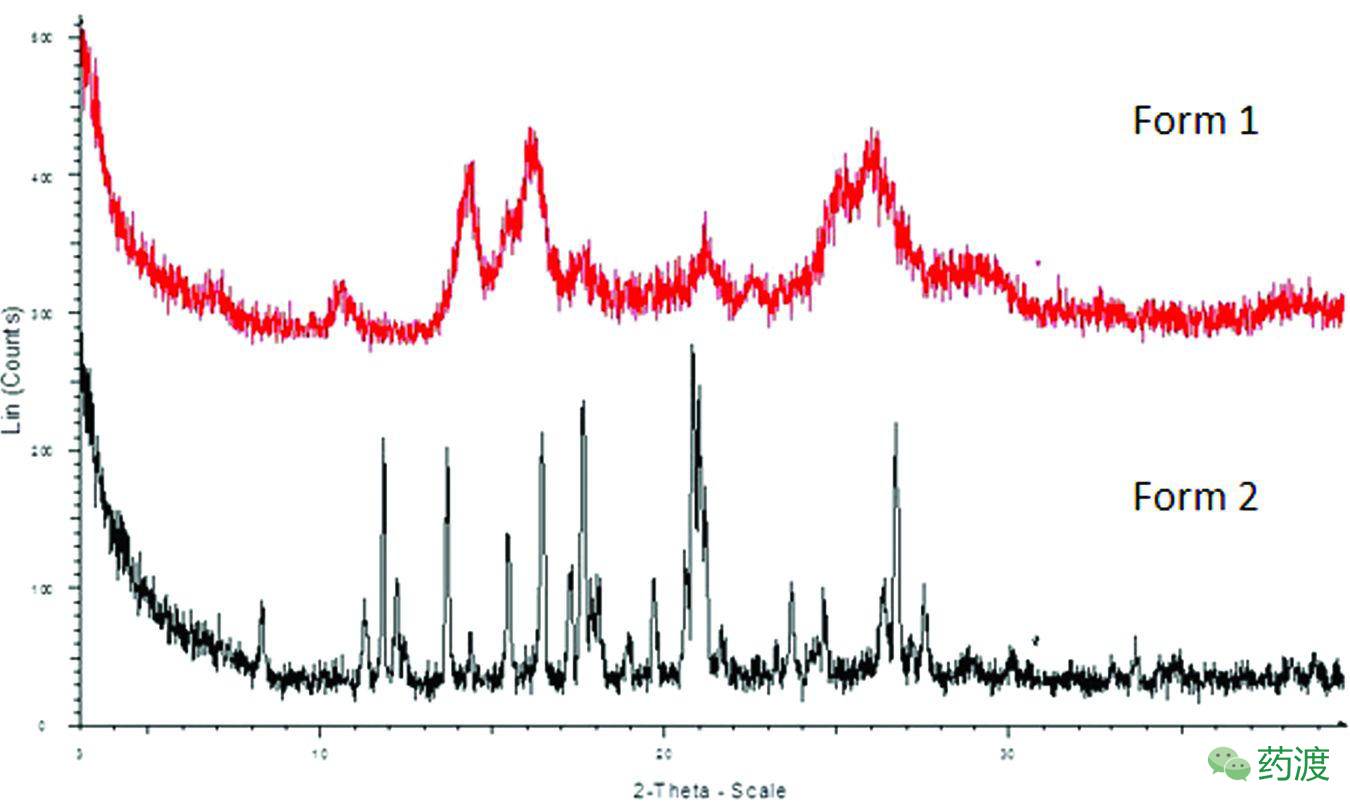
晶型I和晶型II的XRPD图谱
4.总结
经过以上的设计、优化,从4-氯-2氨基苯酚
19
出发,能以12%的总收率制得API。最终API的化学纯度和光学纯度都能满足临床前毒理研究、制剂处方工艺开发和临床I期试验。
具体的制备工艺如下图所示:
随着临床试验的进行,所需的API数量越来越大,现有的中试工艺路线难以满足。研究人员再次对工艺路线进行审视,决定从以下几个方面进行优化:
①手性拆分这一步收率很低,并且还需要进行后续纯化,对整个路线的收率影响很大;
②在工艺路线的早期使用硫光气,对环境和实验人员非常不友好;
③在制备酰胺的这一步,需要提前将胺游离出来,同时相应的羧酸需要转化成酰氯,操作流程复杂。
1.硫光气的替换
使用乙基黄原酸钾与4-氯-2氨基苯酚
19
反应,得到化合物
20
。在巯基的氯化这一步,将二氯甲烷替换成四氢呋喃,在胺化这一步,使用碳酸钾替换三乙胺,在aza – Michael加成这一步,用催化量的氢氧化钠替换早先的DBU。这些试剂和物料的变换能有效的减少废弃物和增加收率。





