如上篇笔者所述(工艺变更在路上:Herceptin多批次发生质量参数偏移),基于科学技术的不断进步,生产条件的持续改变,政策法规的升级,临床和市场需要的满足,生物制品的变更将贯穿于药物研发、生产和流通的整个产品周期中。
抗体药物的经过30多年的发展后,生产工艺已经取得突飞猛进的进展,平台化的工艺已经基本可以满足药物的安全性和稳定性。特别在90年代初期,单抗药物不仅基于免疫原性问题备受质疑,同时相对落后的生产工艺也很难以满足药物的商业化生产。回过头来看,今日抗体药物能够跨入千亿美元市场,不得不感谢那个年代不同领域的研发人员的不懈努力。本文带来的正是首个TNFα单抗药物Remicade上市前后进行一系列生产工艺变更实例,旨在为步履不停的工艺变更之路提供一些参考。
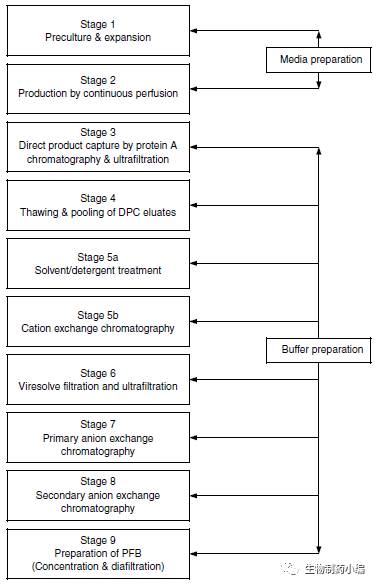
▲ Remicade早期原液生产工艺
早期的Remicade原液生产工艺共分为9步,第1步为细胞的复苏和初步扩增,第2步为大规模的连续灌流细胞培养生产。第3步为细胞培养的澄清和Protein A的亲和纯化,经过初步纯化的产品在进行下一步纯化前先进行冷冻储存。进入第4步,第3步的冷冻样品在室温进行融化后,进行pH调节和过滤。随后在第5步a中,先使用S/D法对产品进行病毒灭活,第5步b使用阳离子层析以去除S/D试剂。第6步使用切向流技术进行病毒的过滤,并对样品进行一定浓缩。第7步和第8步分别使用流穿和结合模式的阴离子层析,以分别去除DNA和病毒以及抗体多聚体和培养基相关杂质。最后的第九步为样品进行浓缩并换液至初步的成品制剂配方溶液中。
临床的开发目的为快速推进候选分子前进,而工艺开发通常来讲集中于提供临床前毒理和临床1期试验的样品。而当时的工艺很可能不能满足商业化的要求,所以生产工艺通常在临床1期试验后会首次进行优化改进。而随后考虑工艺的稳定性、安全性和成本,生产工艺在临床2期、3期甚至BLA前连续进行一系列的工艺优化改进。下图为Remicade在整个临床开发过程中不同阶段进行一系列工艺变更,涉及细胞株、细胞培养工艺、纯化和制剂等各个生产方面,同时为了保证这些变更对产品的质量和有效性没有影响,研发企业进行了一系列的比对研究实验。
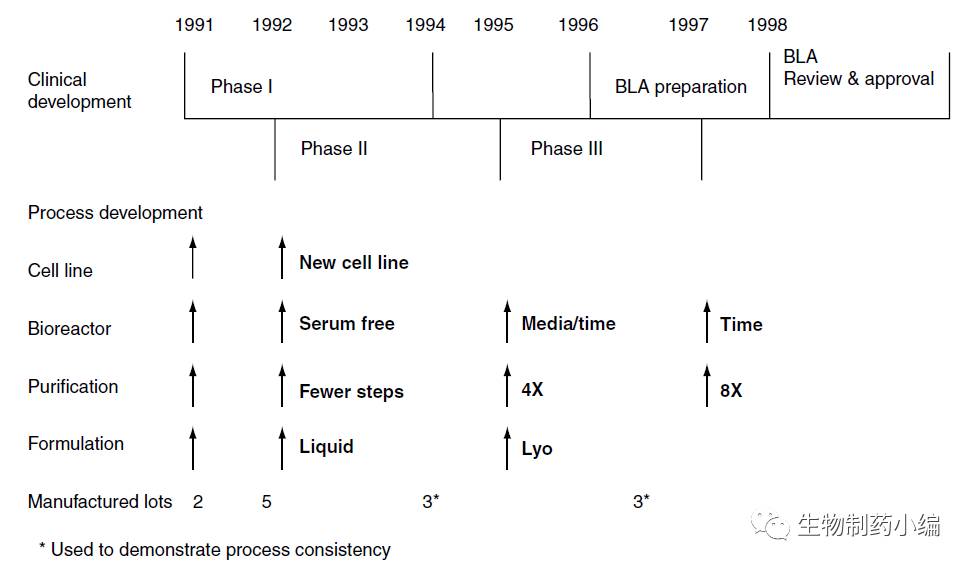
▲ Remicade临床阶段工艺变更之路
1992年,1期临床试验后,Remicade彼时被FDA授予克罗恩病的快速评审资格,为了适应临床的开发进程,工艺开发和生产的相关时间表也进行了调整,一系列相关工艺也进行了优化。
为了提高反应器阶段的生产效率,工艺进行了几个地方的改进。进过细胞株的克隆筛选,原始克隆在方瓶中的表达量从小于9 ug/ml提高到了92 ug/ml,提高了十倍之多。细胞培养基在减少了动物来源组分浓度的同时,单个细胞的药物生产能力提高了2倍。生产反应器培养阶段,连续式灌流维持时间从45到50天提高60天。
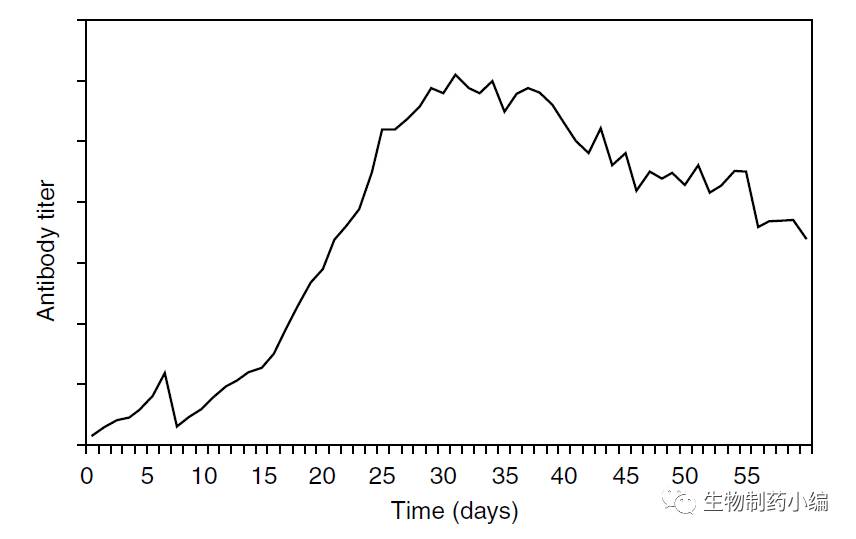
▲ 优化后的灌流培养工艺
上游生产工艺经历以上优化后,下游工艺相应面临更大的挑战。由于产品浓度较低,1期原始工艺中有多步超滤换液步骤,2/3期中均取消了这些步骤。为了提高工艺通量和填料的利用率,离子交换层析的上样量调高了3到4倍。同时为了提高分离效率和产品纯度,也相应的更换了填料,采用高性能的树脂和更小的微球。

▲ 质量比对相关检测项目
随后,在1995年完成2期临床试验后,为了进一步适应临床的需求,进一步进行了以下的工艺变更:
继续优化细胞培养基的配方
进一步提高生产反应器的培养时间
下游纯化工艺放大了4倍
剂型由冻干粉针剂该为注射液
1996-1997年,顺利结束3期临床试验后,Remicade仍然进行了一定的工艺变更,Remicade的剂量由250 mg减少至100 mg,通过理化参数、体外体内生物学活性以及药物动力学试验与临床试验样品完成了比对研究,证明了相似性。
然而上市之后,完成工艺验证不久,为了供应链的可靠性,Remicade又紧跟着进行了一系列的批准后工艺变更。变更较大的为第三步的亲和层析,早前时候该步是下游工艺的限速步骤,原工艺是两根直径为60cm的Protein A层析柱。为了提高这一步的效率,当然增加层析柱的数量使用线性放大的方式更为简单,但从产品质量、法规、成本、时间和人力方面进行了多方考虑,选择更换至更大的直径为80 cm的Protein A层析柱,如下图所示, 层析前后相应过滤和超滤工艺也进行了线性放大(溶液体积增加了1.8倍)。变更后,从实验室规模到生产规模都跟原工艺进行了对比,相关质量参数和工艺参数都较为一致。
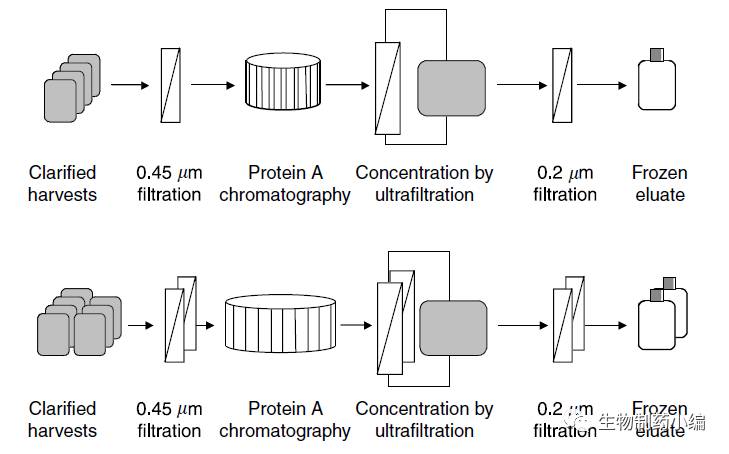
▲ 下游亲和层析工艺变更前后对比
除了以上的跟工艺相关的验证,此变更在生效前还补充以下研究:
而不同监管机构中,此工艺变更的申报路径也是不一样的:
无论哪种监管机构,都需要提高以下资料:
计划变更工艺的描述
计划变更工艺的目的
执行的工艺变更工作的总结
五个连续亲和层析验证批中中间样品检测结果
工艺变更前后杂质去除能力比较
工艺变更前后亲和层析工艺性能参数比较
五个连续亲和层析验证批相应的原液批放行数据
Remicade上市后亲和层析的规模放大的工艺变更工作历时17月,变更得到批准后,生产能力得到显著提升。
随着上市后全球供应链需求的不断增加,荷兰莱顿(Leiden)工厂的产能已很难满足,因此Centocor公司于1997年,在美国宾夕法尼亚莫尔文镇(Malvern)新建一座工厂,整个工艺流程和莱顿一样都为9步,只是Malvern工厂的规模提高了两倍,同时自动化程度也更高。生产反应器体积提高至1000L,同时下游相应病毒过滤工艺的规模也提高了。

▲ Malvren外源spinfilter的1000L灌流反应器
基于更大的反应器规模,事实上在Malvern工厂的9步工艺中每步都进行了相应的规模放大,有的为线性放大,而阴阳离子交换同样基于成本等多方面的考虑更换至更大直径的层析柱。
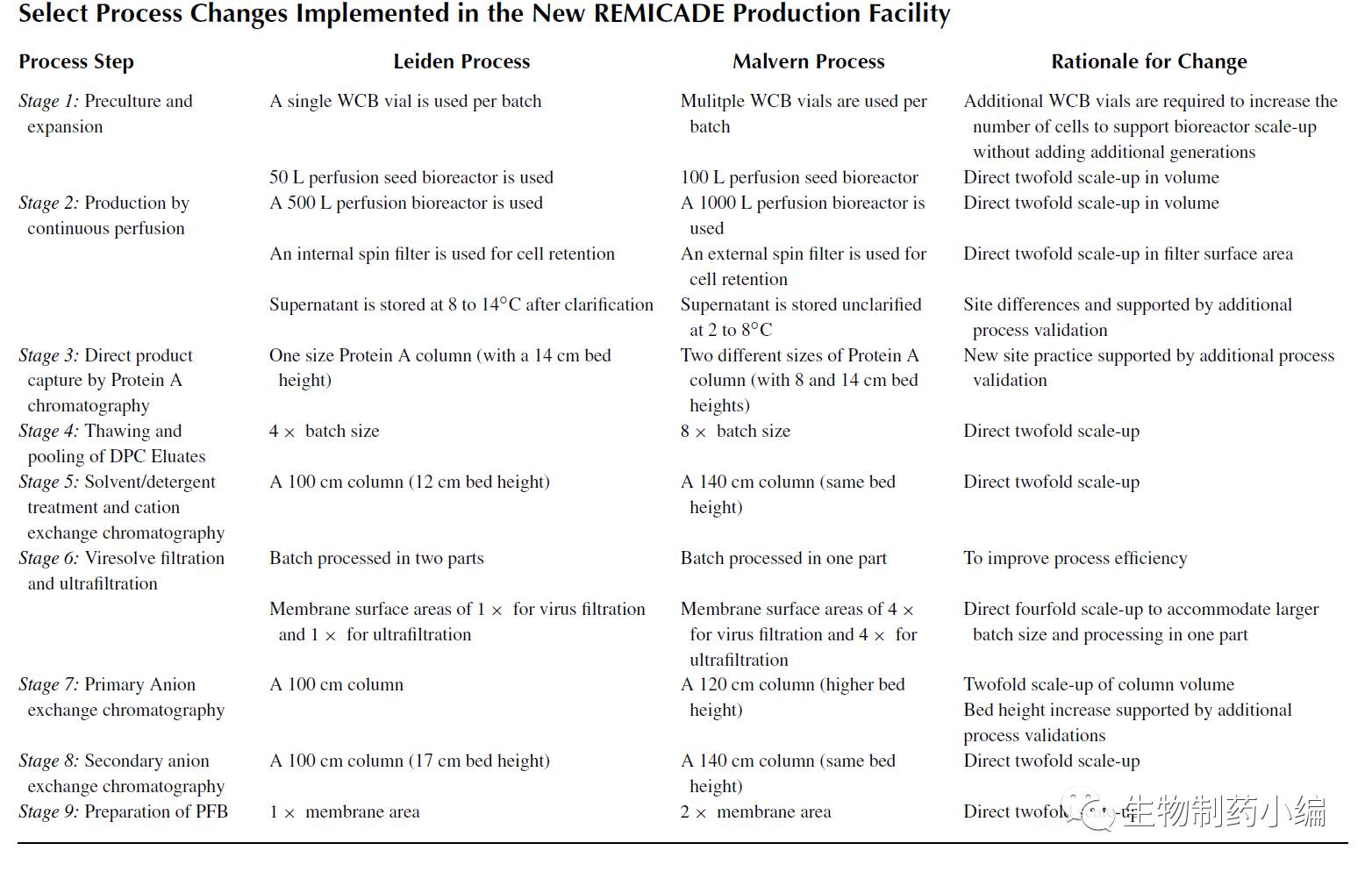
▲ Malvren工艺变更内容
Malvern工厂建成后,2000年10月新厂完成了连续4批的工艺验证批,与同时期(2001年)Leiden工厂生产的三批以及3批Leiden工厂在1997-1998年的验证批均进行了比对性研究,结果显示三者之间质量无明显差异。

▲ Malvren工艺变更验证方法内容
相关的工艺验证工作完成后,Malvern工厂于2002年被FDA认证批准,次年被EMEA认证批准。2002至今,Leiden和Malvern两个工厂变更工艺数次。截止到2007年,成品生产地也从最初一个(Parkdedale)扩充到5个。基于频繁的工艺变更,彼时Centocor也创造了首轮审评批准的记录。他们总共向FDA递交50多项Remicade相关的工艺变更申请(均是重大变更和中度变更),除了1项以外其余均在首轮审评得到批准。
根据CFR 21. § 314.70中相关条款,上市后的变更主要分为以下三种:
1. 重大变更
这种变更属于会引起药物的有效性或者安全性的重大变化,这种情况下需要按照Prior Approval Supplement进行申请,这种变更需要得到FDA的批准后才可以上市销售变更后的产品。
2. 中度变更
这种变更为中度变更,根据情况又可以分为两种,CBE30 和CBE0。CBE30将变更资料递交至FDA, 若FDA没有进一步意见,30天之后即可上市销售变更后的产品;CBE0将便跟资料递交至FDA的同时就可以上市销售变更后的产品
3. 微小变更
这种变更对产品的安全性和有效性的影响很小,可以采取Annual Reports的方式递交,不需要FDA批准,应当属于告知FDA信息的一类变更。
而关于怎么分类相关的变更,则依据FDA于2004年颁布的Changes to an Approved NDA or ANDA的指导原则,企业可以对照自身的变更情况进行相应类型的变更。
此外,为了有效地保证供应链以满足临床日益增加的需求,Centocor有效地利用可比拟变更方案(Comparability Protocol,CP)作为常规的申报策略,该方案可以允许降级为更小的报告类型。Malvern厂址2007年前共申请了7个CP,除了1个没有得到批准,其余6个都被同意以更小类别的工艺变更类型进行验证。
CP是一个明确、详细的书面计划,用于评估特定的CMC变化对一个特定药物产品质量的影响。通常一个CP方案定包括:1、该方案所涵盖的变更的说明;2、执行的检验和研究,要使用的分析程序,和要达到的认可标准,以证明变更后没有不良影响;3、提出报告类别(一般比普通类别低1级)。CP多用于减少实施变化的报告类别,从而是变更后的产品尽早进入市场。它适用于以下变更情况:
综上所述,基于FDA的PAS和CP机制对于上市后的原液(API)产地变更的具有明显的优势,可以极大的减少药企工艺变更进行的研究内容和等待批准时限。

▲ API上市后产地变更审评制度比较
本文只列举2007年前公布的Remicade临床开发及上市后工艺变更记录,或许从内容上已经有些陈旧,但对于Remicade特定例子来看,仍具备一定的代表性,几乎涉及到了抗体药物生产的各个方面,笔者认为仍具有一定的指导性。
特别是对于今日整个重心已经转移到临床后期的中国生物制药来讲,细胞株的更换、工艺的优化和产地的变更等一系列CMC工艺变更或许正在进行,这不仅仅药企的本身的工作。如何形成有效的沟通机制,如何进行科学且经济的监管机制,如果有效减少工艺变更涉及的研究和审批时间,也应该是中国监管机构需要作出的努力。
工艺变更在路上:升级不断,变更不止。
参考资料
欢迎加入小编团队成为小编一员
请加小编微信号:wuwenjun7237
如有技术解读、行业洞见愿意分享
欢迎投稿到小编邮箱:[email protected]

版权为生物制药小编所有。欢迎个人转发分享。其他任何媒体、网站如需转载或引用本网版权所有内容须获得授权且在醒目位置处注明“转自:生物制药小编”。

坚持原创、坚持专业
欢迎关注生物制药小编
投稿信箱:[email protected]
小编团队现有13位成员:
Armstrong、医药局外人、Fairy、Jone、
东胜西牛、Alpharesearcher、MT、百草、
Irene、北望、蛋白工人、At.Zhou、Julia
欢迎有共同兴趣的朋友加入








