双碳目标的提出为以氢能为燃料的清洁、高效的燃料电池技术带来了机遇。然而,“Pt原子经济性”的问题成为其商业化面临的一大挑战。近年来,碱性膜燃料电池技术(AEMFCs)以其突出的成本优势而受到广泛关注,有望大幅度缓解Pt依赖性这一“卡脖子”问题。但是其阳极端氢氧化反应(HOR)动力学较酸性介质慢2个数量级,现有的催化剂活性和稳定性依然堪忧。特别是稳定性,详细的催化剂衰退机制尚不明确,严重影响了催化材料设计策略的提出。
为此,
华中科技大学王得丽教授课题组
在
Journal of the American Chemical Society
期刊上发表了题为“Pseudo-Pt Monolayer for Robust Hydrogen Oxidation”的文章(DOI: 10.1021/jacs.2c11907)。针对稳定性的问题,该团队先从商业Pt/C入手,提出两段式的衰退机制,即:1. 电位低于0.5 V vs RHE时,Pt原子催化碳载体氧化生成CO,CO*物种随后反溢流至Pt表面导致催化剂毒化,电化学活性面积降低。2. 电位高于0.5 V vs RHE时,溶液中的OH*物种促使Pt纳米晶表面的Pt原子脱离本体,活性位点锐减。这一两段式衰退机制的提出不仅对于碱性HOR,甚至有望应用于碱性介质中的以Pt/C为商业催化剂的多种催化反应的活性衰退分析,从而有利于提出可靠的稳定性提升策略。依据此衰退机制随后提出一种“缓冲介质”策略,在碱性介质中拥有强壮稳定性的IrPd内核表面外延生长赝Pt单层结构,形成一种新型的复合结构催化剂(PmPt@IrPd/C),该催化剂能够结合内核和外壳的高稳定性和高活性,实现兼顾提升催化活性和稳定性的目标。研究表明,其在0.5 V vs RHE的电位下能稳定工作50,000圈,在1.0 V vs RHE的高正端电位下仍能稳定工作20,000圈,均明显优于商业Pt/C。更重要的是,以PmPt@IrPd/C为阳极催化剂装配的碱性膜燃料电池在超低的Pt用量下,功率密度可达1.27 W cm
-2
。该研究成果为碱性膜燃料电池中高活性和高稳定性的催化剂开发提供了理论依据和设计思路。
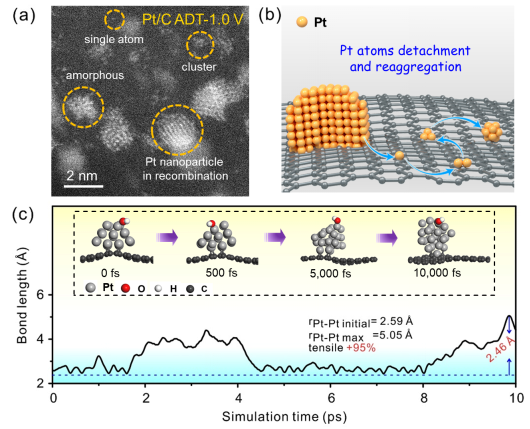
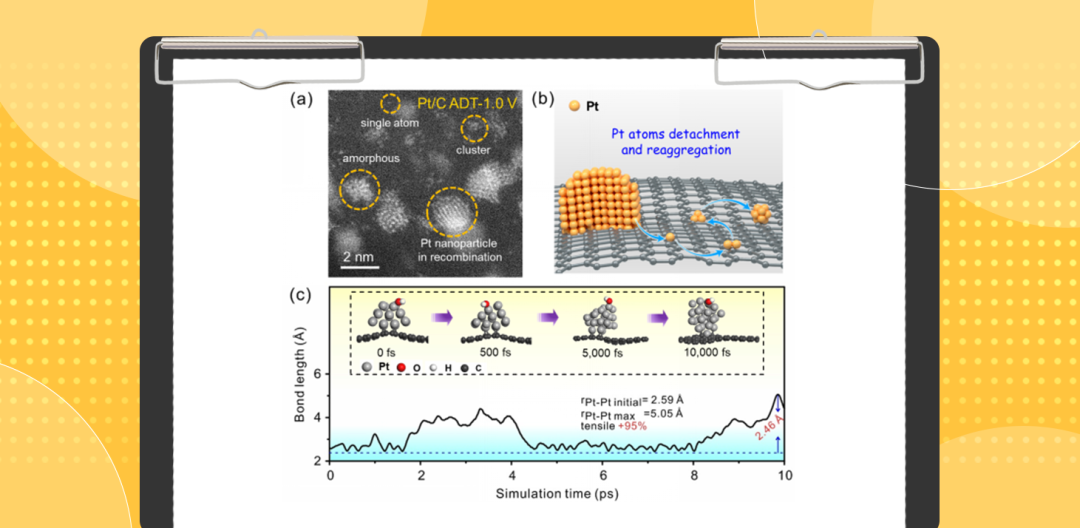
图1
. Pt/C催化剂在0.1 M KOH溶液中的HOR性能评估。(a) Pt/C催化剂在1.0 V正端电位稳定性测试后的HAADF-STEM图。显然,老化后的Pt/C样品中出现单原子、团簇、纳米颗粒等多种结构。(b) 提出的Pt/C催化剂衰退机制,表面的原子从Pt纳米晶表面分离并重组为单原子和纳米团簇。(c)在10 ps的分子动力学模拟中,表面和亚表面Pt原子之间Pt–Pt键长的变化趋势。插图显示Pt/C模型受表面吸附OH*物种拖曳效应影响的结构演变趋势。
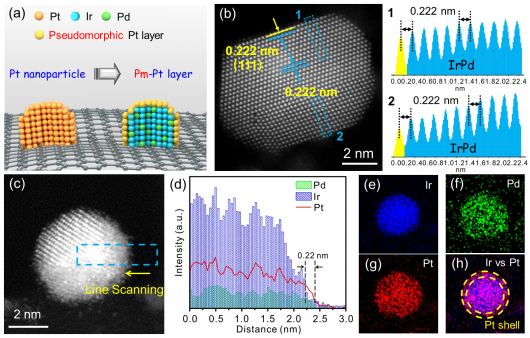
图2
. PmPt@IrPd/C催化剂的结构设计理念与相关结构表征。(a)从Pt 纳米颗粒到PmPt@IrPd核壳结构催化剂的设计理念示意图。(b) PmPt@IrPd纳米颗粒原子级分辨率的HADDF-STEM图,其中黄色和蓝色实线之间的距离为PmPt层和IrPd核之间的晶面间距,IrPd核的晶面间距用双蓝色实线表示。图1和 2分别表示PmPt@IrPd 纳米颗粒中相应区域1和2中的强度曲线图。(c,d)和(e-h) 分别表示单颗PmPt@IrPd纳米粒子的元素线扫图和EDX元素分布图。很显然,Ir和Pd均匀地分散在内部,Pt分布在表面,且测量的PmPt壳层的厚度约为0.22 nm。
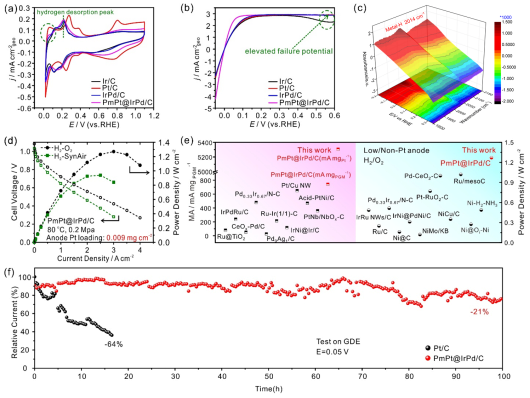
图3
. PmPt@IrPd/C催化剂的HOR电催化性能分析。(a)所制备催化剂在Ar饱和的0.1 M KOH溶液中的CV曲线图。(b)所制备催化剂在H
2
饱和的0.1 M KOH电解液中的HOR极化曲线图。(c)在0.1 M KOH溶液中,PmPt@IrPd/C催化剂于0.35V至-0.20 V电位范围内的原位ATR-SEIRAS谱,图中显示PmPt@IrPd/C中的M–H带位于2014 cm
–1
波数处。(d) PmPt@IrPd/C作为阳极催化剂组装出AEMFCs的极化曲线和功率密度曲线图。(e) PmPt@IrPd/C的质量比活性和装配的AEMFCs的电池性能与先前报道的别类催化剂相比较。(f)在GDE上利用计时电流测试技术对Pt/C和PmPt@IrPd/C的稳定性进行比较。
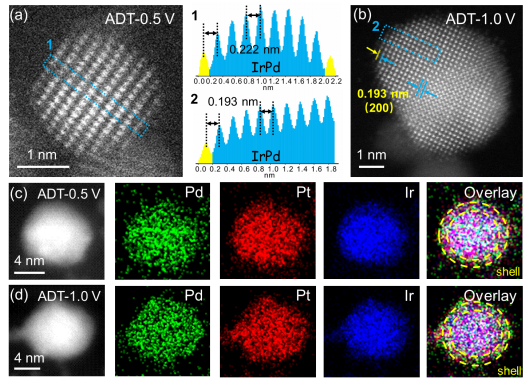
图4
. PmPt@IrPd/C催化剂的稳定性结构表征。分别经过(a) ADT-0.5 V和(b) ADT-1.0 V测试后, PmPt@IrPd/C催化剂原子级分辨率的HADDF-STEM图。图1, 2为(a,b)中PmPt@IrPd纳米颗粒中相应区域1和2的强度曲线图。PmPt@IrPd纳米颗粒分别经过(c) ADT-0.5 V和(d) ADT-1.0 V测试后的元素分布图。
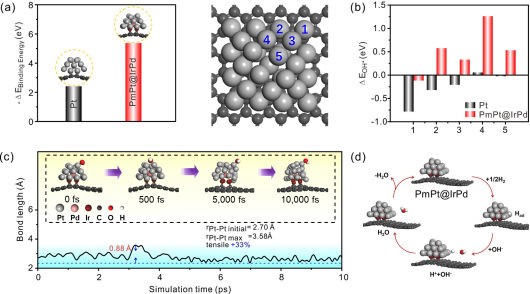
图5
. PmPt@IrPd/C的密度泛函理论计算和分子动力学模拟。(a) Pt和PmPt@IrPd模型与碳基底的结合能。(b) OH*物种在Pt和PmPt@IrPd模型5种可能的吸附位点上的结合能,左侧为相应吸附位点构型的俯视图。(c)在10 ps分子动力学模拟过程中,表面和亚表面Pt原子之间Pt–Pt键长的变化趋势。插图显示PmPt@IrPd/C模型受表面吸附OH*物种拖曳效应影响的结构演变趋势。(d) PmPt@IrPd/C模型上HOR的机理示意图。









