上一期我介绍了三种常用集群作业调度系统的基本用法,本期主要介绍一下我如何安排目录结构以及利用作业调度系统实现相似任务的自动提交及并行计算,供大家参考,欢迎大家留言讨论。
目录结构安排
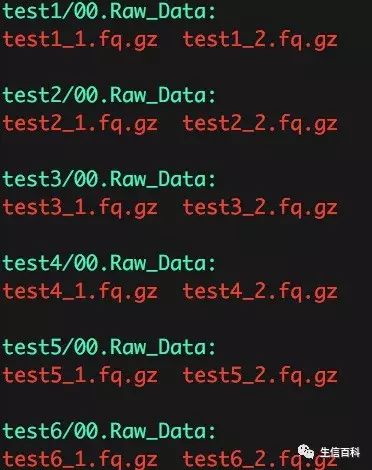
我以 6 个个体的测序数据为例来演示一下我如何安排目录结构,原始数据 (双端测序) 如下图所示:

 我通常以每个样本的名称建立文件夹,然后在每个样品下面创建各计算过程的子文件夹 (如: 00.Raw_Data, 01.FastQC, 02.Clean_data 等),具体的实现代码如下:
我通常以每个样本的名称建立文件夹,然后在每个样品下面创建各计算过程的子文件夹 (如: 00.Raw_Data, 01.FastQC, 02.Clean_data 等),具体的实现代码如下:
for i in $(ls *_1.fq.gz)
do
name=${i%_*}
mkdir ${name}
cd ${name}
mkdir 00.Raw_Data
mv ../${name}_1.fq.gz ../${name}_2.fq.gz 00.Raw_Data
cd ..
done
我采用的思路是提取正向的序列名 (ls *_1.fq.gz),去掉后面的_1.fq.gz (${i\%_*}) 得到样本的名称,以样品名称建立文件夹并进入此文件夹,创建 00.Raw_Data 文件夹后将原始数据移动到此文件夹;
整个过程使用 for 循环完成,即使有再多的样品也能短时间内完成。
运行结束后,目录结构将如下图所示:
任务的自动递交及并行计算
在上述的目录结构基础上,利用集群作业调度系统,可以很方便的实现任务自动递交及并行计算。以 FastQC 的运算为例:
cd $PBS_O_WORKDIR
mkdir 01.FastQC
path=$(pwd)
fastqc --noextract -t 2 ./00.Raw_Data/${path##*/}_1.fq.gz ./00.Raw_Data/${path##*/}_2.fq.gz -o ./01.FastQC
假设 test1, test2, test3, test4, test5, test6 位于 data 文件夹下,接下来我们在 data 文件夹下新建一个名为 fastqc.sh 的文件,将上述 Torque 作业系统的运行代码写入 fastqc.sh 并保存,然后运行下面的代码:
echo ./test* | xargs -n 1 cp fastqc.sh
rm fastqc.shfind $PWD -type f -name "fastqc.sh" > run.txt
for i in $(cat run.txt)
do
cd ${i%/*}
qsub fastqc.sh
done
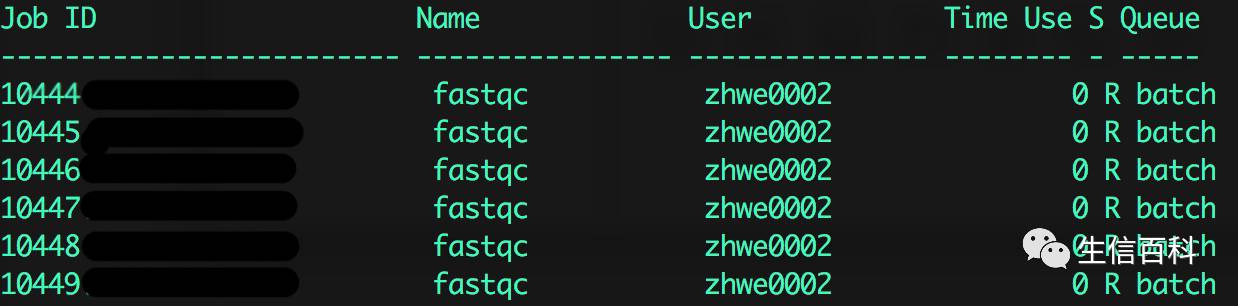
运行 qstat 会看到所有任务都开始运行了:
由于每个文件夹都是以样本名称命名的,我们可以用
变量
来代表样品名称 (path=$(pwd); ${path##*/} 即为样品名称),这样写的脚本适用于
无数个
目录结构相同的样本;
将脚本拷贝到每个样本的目录下,用 find 找到脚本,之后可以用 for 循环完成脚本投递任务;
理论上,只要有足够的计算资源,可同时分析成百上千个样本。
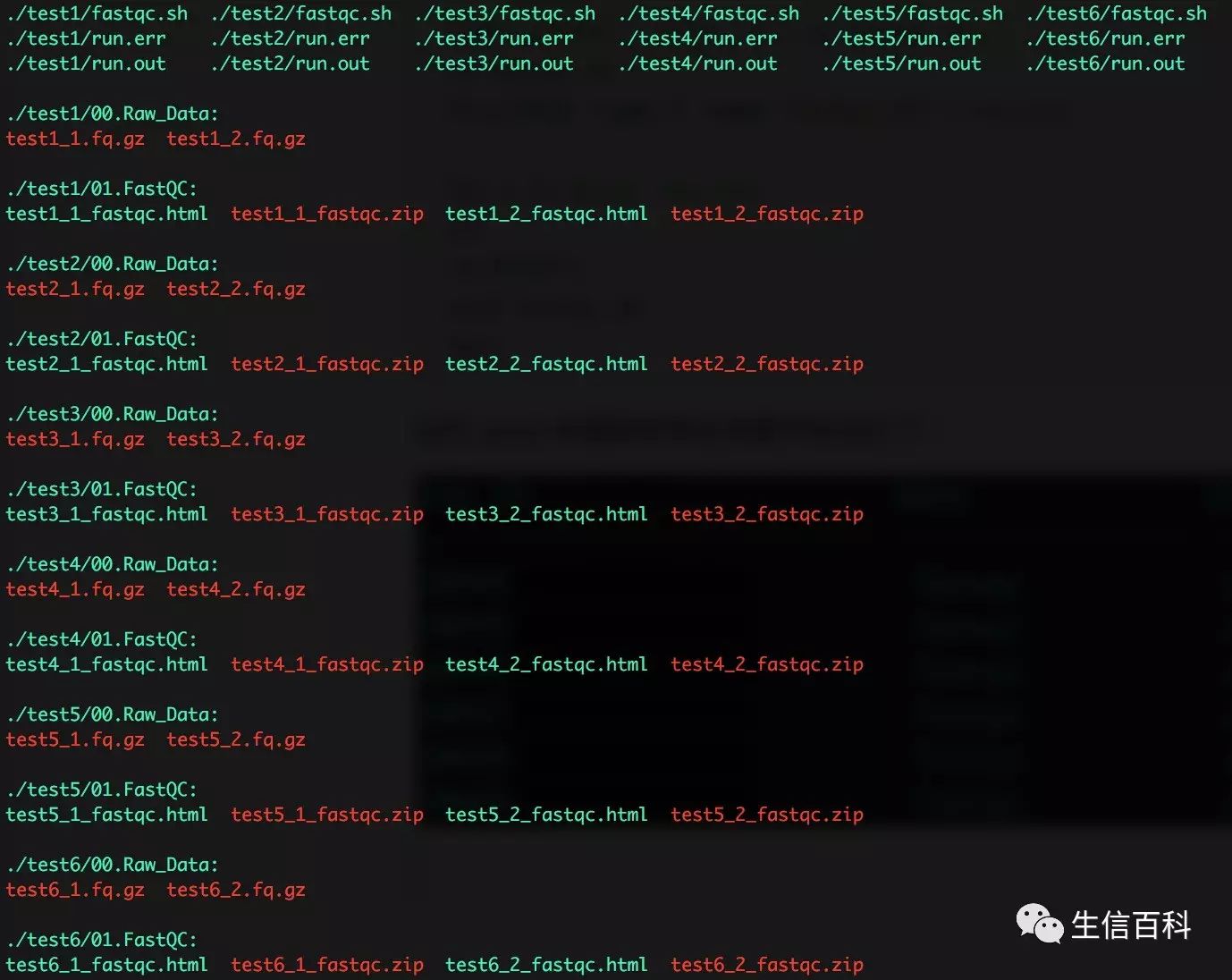
脚本运行结束后,最终的目录结构如下:
往期回顾: