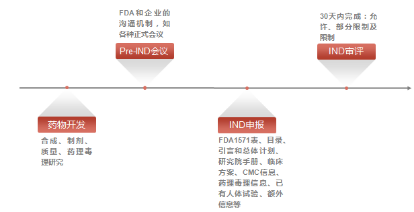
( FDA 的 IND申报审批流程图)
新药临床试验申请(Investigational New Drug Application,IND)是美国 FDA 对尚未上市审批、需要进行临床试验的药物的许可。另外,美国联邦政府禁止未获 FDA 批准的药品在美国进行跨洲际间运输和分发,因此,对于主办者将药品运输到外州进行临床试验的情况,IND 也是申请对这项法律条款的豁免权。
每年 FDA 都会收到很多 IND 申请,中国企业向 FDA 申请的 IND 数量也颇为可观,但是中国企业大多数 IND 申请是在国内已经开展临床试验甚至上市后,或者在澳洲启动临床试验后,再向 FDA 申报 IND,直接向 FDA 申报 IND 开展首次临床试验的较少。
那么,什么情况下需要提交 IND 申请呢?在新药或生物制剂完成临床前研究,准备开展临床试验时,需要提交 IND 申请。亦有在已获批药物或生物制剂发生新的适应症、重要标签变更、以及剂型、给药途径或患者群体(如,儿童、性别)的变更时也需要提交 IND 申请。
在提交 IND 申请前,申请人需对药物进行一系列临床前研究,以证明该药物在人体试验中使用是安全的,同时申请人还应确定该药物是否确有药理活性。当临床前研究证明该药品可进一步开发为上市产品时,申请人应在有限的早期临床试验中收集必要的数据和信息证明该药物对人体不会产生不合理的风险。一旦这样的药品数据确定(或与动物试验数据相对应)且生成报告,申请人即可开始准备 IND 申请。
IND 申请前准备所需时间与产品的特征信息、非临床试验和临床试验数据是否准备充分有关。许多首次提交 IND 的申请人低估了提交一份设计完善且操作性强的 IND 所需的时间,事实上,从发现一个有临床资格的候选药物起,便可能花费两年甚至更长的时间准备一份完善的 IND 申请。理想情况下,申请人应对认真学习 FDA 官网上 IND 相关的指导文件和表格,以更好的设计产品满足要求。
一般而言,IND 申请包应包括三大层面的信息:
1)动物药理/毒理学研究:临床前数据应充分证明该药品对于人体的首次试验是安全的。主要涉及药效、药代、毒理学等研究,核心内容是安全性评价,如 GLP 毒理试验。FDA 根据关键毒理研究资料,观察安全窗,决定是否接受临床试验方案。
2)CMC 资料(生产信息):包括药物产品的组成、生产工艺、稳定性和生产控制等信息,目的是充分证明生产商可以连续生产和提供质量稳定和统一的测试样品。
3)临床试验方案和研究员信息:FDA 决定是否允许开展临床试验的标准是评估对受试者是否有不可接受的风险。在美国提交的临床方案是包括很多细节的可执行的方案,需要和 CRO、医生进行多次沟通交流方能确定。此外,临床试验的监督管理者(专业人员或医生)应具有从事临床试验相关工作的资格。还包括,受试者的知情同意书,以及该测试应在机构审查委员后(IRB)的监督之下且遵守 IND 条例的相关规定。
在申请人提交 IND 后,FDA 的项目监管经理作为监管联络人将 IND 申请转交给审查小组。通常审查小组由化学家、药理学家/毒理学家、临床医生、统计学家和药代动力学家组成,若药品中包括医疗设备,还应咨询设备和放射健康中心(CDRH)的审查人员。
在 FDA 收到 IND 申请后,必须在 30 天内通知申请人是否可以进入临床,在此期间,FDA 需对 IND 安全性进行审查,以确保受试者不会受到不合理的风险。由于 FDA 可能会要求申请人提供额外的详细信息或声明,因此建议申请人组织企业内部的 IND 工作小组在 30 天内进行充分地讨论,以便及时解决 IND 的相关问题。
假如申请人收到 FDA 的通知(通常是以电话或电子邮件的形式通知,且随后 FDA 会发送 「允许临床(Safe to Proceed)」通知),则可以进行临床研究,同时该 IND 为「激活」状态。
若 FDA 认为临床试验存在安全风险,有权力向一项研究或者研究机构发出临床试验暂停(Clinical Hold)通知,并告知理由。临床试验暂停可以是全面或者部分的暂停。FDA 已经颁布了对涉及药品和生物制品研究发出临床实验暂停通知的条例(21CFR312.42)。
通常申请人需在 30 天内针对 FDA 提出的问题进行资料补充并提交书面的完整回复,一旦收到对临床试验缺陷的完整回复,FDA 将在 30 日内对提交内容进行重新严格的审评,如果 FDA 认为问题已经解决,便会通知申请人临床试验可以继续开展(Off Hold)。
通常导致「临床试验暂停」的原因有以下几点:
1)证明药品安全性的信息不足;
2)临床试验批次的杂质不合格或杂质信息表征不充分;
3)主细胞或工作细胞库或病毒库的研究不充分;
4)药品在试验过程中不稳定;
5)毒理学研究未定义「无可见不良作用水平」(NOAEL);
6)非临床试验数据不能支持最大人体剂量的界定;
7)临床方案设计不符合规定的目标;
8)临床研究设计缺乏必要的安全监测,以及/或「试验中止规则」未被定义或不充分。
9)缺乏研究员声明(FDA 表格 1572)。
一旦某 IND 处于「激活」状态,它将成为一个可更新文档。作为该 IND 的申请人,需随时对 IND 进行更新,如研究数据、新协议文件,协议文件修订、安全性报告、新技术信息、新有效性数据以及其他的相关信息等。大多数的文件更新是通过提交 IND 修订完成,如以下类型:
1)方案修订 Protocol amendment (21 CFR 312.30);
2)信息修订 (21 CFR 312.31);
3)安全报告 (21 CFR 312.32);
4)年度报告 (21 CFR 312.33);
5)对 FDA 信息请求的回复。










