片剂的性状检查是一个重要的物理稳定性考察指标,通过目测法观察片剂表面或除去包衣后片芯的颜色是否符合规定。此外,在片剂的生产环节,如压片、包衣工序也会对其外观进行检查,如片剂表面应无黑点,花面等现象。当片剂性状出现问题后,如何调查分析呢?
本文首先通过一个案例,介绍稳定性考察中片剂出现黑点后的分析思路和解决办法,其次谈一下作者关于类似问题的一些思考,仅供研发同仁参考。
一、案例介绍
【问题描述】
片剂在长期(25˚C/60%RH) 和加速(40˚C/75%RH)条件放置3个月后,在加速条件下出现黑点,变化如下图所示:

【问题调查】
经调查,片剂在放行和生产过程中未见黑点,因此排除了原辅料、生产过程受到污染的可能。经分析,很可能是由于加速稳定性条件下发生了化学变化引起的,因此,继续进行调查。
1、原辅料情况
所有辅料均商业可供,符合USP/EP/JP标准,API在片中含量低为0.1~0.4mg/片。辅料种类如下:
Lactose monohydrate
Hydroxypropylmethylcellulose
Starch 1500 (pregelatinized starch)
Sodiumlauryl sulfate (SLS)
Colloidal silicon dioxide
Magnesium stearate.
2、微生物限度检查
在加速条件下温湿度较高,怀疑可能会产生微生物污染,因此首先把出现黑点的药片进行微生物限度检查,结果表明微生物限度符合规定,说明微生物污染不是引起黑点的根本原因。
3、原辅料相容性研究
由于黑点在产品贮藏过程中产生,因此可能由原料辅料的相互作用产生。因此进行了二元混合试验(辅料/辅料,原料/辅料),并在60 °C 和 40 °C条件下放置6周。每个辅料都采用了3个不同批号,目的是考察辅料批间的差异。片芯和包衣片也进行了放置,考察包衣材料对片芯是否存在影响。
二元混合试验结果表明,在一批SLS中没有出现变色问题,而且,随着温度的升高,药片的变色程度会增加。此外,片芯和包衣片都出现了变色,说明片芯和包衣材料不存在相容性问题。
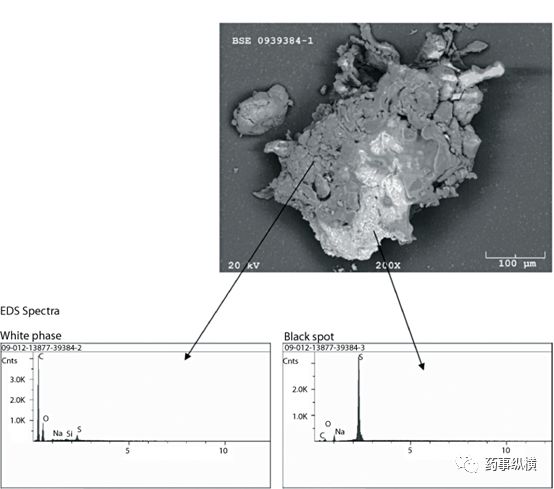
4、图像和元素分析(SEM/EDS)
采用扫描电子显微镜和能谱分析技术(SEM/EDS)对药片的变色区域(黑点区域)进行检查,在图谱中,白色区域代表片剂中的黑点。EDS图谱中发现药片黑色区域中含有硫元素(S)和钠元素(Na)。处方中含量这两种元素的辅料是SLS,因此判断SLS与黑点的形成有关。
5、不同来源SLS的研究
采用两个不同批号的SLS的4个样品进行研究。批号A是加速试验中产生黑点的批次,批号B来自另一个不同的供应商。这两批样品在室温和60°C条件下放置4周,结果见下表。
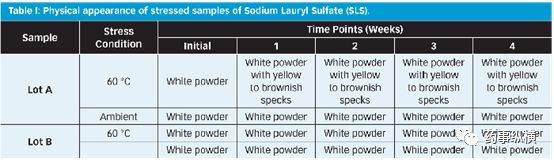
结果表明,批号A的SLS样品中出现变色,而批号B的SLS样品中没有出现变色。由于这两批SLS均符合USP/EP/JP,因此继续研究二者之间存在的细微差别。
质谱(MS)分析:
将批号A和B的溶液直接进样,进行MS/检测,同时采用正离子和负离子模式进行分析。
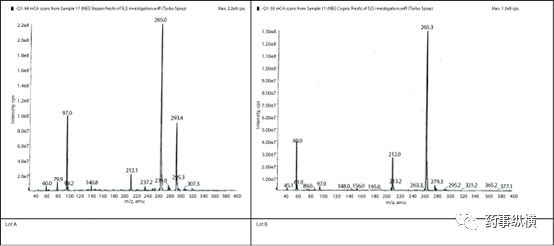
上图中的负离子模式质谱图表明,m/z=60.0和212.0峰为空白峰,m/z=265为SLS (C12H25SO4)峰。此外,有两个大峰(m/z=293和97)仅在批号A的SLS中发现,这两个峰归属为十四烷基硫酸(C14H29SO4)和残留的硫酸 (HSO4)。正离子模式峰很多,很难分析,但是批号A明显比批号B峰多。综合正负离子模式的分析可见,批号B的纯度比批号A高,另外值得注意的是批号A的硫酸峰在批号B中没有出现。
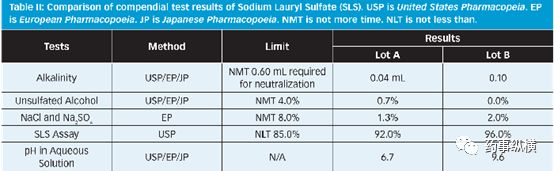
药典方法检测结果对比:
将两批SLS的QC放行结果进行对比,结果表明,二者虽然均符合USP/EP/JP标准,但是批号A中未酯化醇的含量高于批号B,含量测定结果低于批号B。此外,批号A的碱度和pH值均比批号B低,进一步说明批号A中含有一定量的硫酸。
6、结果讨论
根据上述一系列的调查分析,药片的变色与处方中SLS中的杂质紧密相关。SLS目前有三种生产工艺:
工艺1:SO3 /airprocess
Step 1 C12H25OH + SO3 → C12H25SO4H
Step 2 C12H25SO4H + Na2CO3 → C12H25SO4Na + CO2 + H2O
工艺2:Oleum (H2SO4 /SO3 )process
Step 1 C12H25OH + H2SO4/SO3 → C12H25SO4H + H2O
Step 2 C12H25SO4H + Na2CO3 → C12H25SO4Na + CO2 + H2O
工艺3:Chlorosulfonicacid process
Step 1 C12H25OH + ClSO3H → C12H25SO4H + HCl
Step 2 C12H25SO4H + Na2CO3 → C12H25SO4Na + CO2 + H2O
月桂醇来自植物,其中会含有少量的C-10、C-14和 C-16同系物,在反应中会转移到SLS中去,由于理化性质相似,这些同系物不认为是SLS的杂质。但未反应完的醇、硫酸、盐(NaCl、Na2SO4)、Na2CO3是主要的工艺杂质,这就是为什么药典标准中规定碱度、氯化钠、硫酸钠、未酯化醇含量的原因。
SLS中的杂质谱和杂质量与工艺与控制水平有关,例如工艺2中就可能有硫酸残留。对于工艺2和工艺3而言,如果能够严格控制反应条件就会有很少的硫酸残留在终产品。
批号A可能由于中和步骤不完全,而导致硫酸残留,这通过检验数据可以佐证。本品在高温条件下,SLS中残留的硫酸与碳水化合物辅料(如乳糖)发生脱水反应,导致了黑点的产生。
二、思考与建议
1、原辅料控制
原料药生产中,起始物料、溶剂试剂、合成工艺、精制方法、生产环境、设备等都可能引入外源性杂质或黑点,包装过程中也可能引入灰尘。原料药本身如果发生降解也可能产生黑点,在研发前期就应该对文献进行检索,对化学性质进行归纳总结。在研究过程中的高温、高湿、光照等降解试验中也应注意观察颜色的变化。
辅料质量是影响产品质量的关键因素,由于生产工艺以及质量控制水平的差异,不同来源、不同批号、不同厂家的辅料都可能存在很大差异。就某一辅料而言各国药典标准往往不一致,在药品注册申报中应注意对所使用的辅料进行各国标准列表对比,分析存在的差异及原因,并结合特定产品的处方工艺制定合理的内控标准,有时候看似增加很多研究工作,但是对产品质量而言可能是一个重要保证。










