作者 l 游方和尚
编辑 l 小卒子
本文为探讨如何判定药物毒理研究中的不良和非不良效应的系列文章的第一篇。本文根据已发表的文献资料和小编之前的相关文章做出的一个简介。希望起到抛砖引玉的目的,引发更多的相关讨论,交流和研究。
导论
药物研发(R&D)是一个漫长,复杂而且昂贵的过程,失败远远多于成功,进入临床I期研究的新药分子大约只有10%最终成功地得到美国食品和药物管理局(FDA)的批准。过去30多年来研发投入的增加并没有显著地转化为更多的药品审批成功、上市,造成了药物研发生产率的差距:即增加投资而没有相应的增加成功率。有许多因素可以解释这种生产率的差距。其中一个原因是由于药物安全性问题导致的失败。特别是由于非临床安全性不足造成的损耗是提高研发生产力所面临的一个主要问题,特别是在药物发现的优化阶段和临床开发的早期阶段。
在开发的新药进入首次临床试验之前,确定人体安全起始剂量是最重要的步骤之一。在理想的情况下,起始剂量应该低,不会对人体造成任何伤害; 但也不能过低,从而减少首次接受人体(FIH)临床试验中接触无效剂量的患者人数。FDA相关指南的重点放在了临床前毒理学研究中用药剂量的未观察到不良效应
水平
(NOAEL)上。然后通过应用适当的比例因子,将NOAEL转换为人体等效剂量(HED)。确定HED后,需要更进一步使用适当的安全系数降低剂量从而得出最大推荐起始剂量(MRSD),以保证起始剂量不会对人体造成毒性。
为了预测人体的安全暴露量,有必要描述观察到药物毒性的边界,即首次观察到不良效应的最低剂量(LOAEL)或未观察到不良反应的最高剂量(NOAEL)。为了确定LOAEL和NOAEL,就必须评估在研新药内在的毒理学特性。这些毒理学数据可用于:1. 确定潜在的危害; 2. 通过比较动物模型中产生这些危害的剂量与已知或估计的人体暴露量,来评估对人体的潜在风险(在特定的条件下)。关键的决定因素是受试人群应避免的不良效应的性质以及其产生和不产生时的人体暴露量。定义这些危害(以及随后的评估风险)的核心是清晰地确定毒理研究中观察到的与治疗相关的变化是否是被测试药物对受试物种产生了不良效应。
由此可见,一种科学上可靠且稳健的安全评估方法要求在定义这些不良反应的特性/特征时保持一致性。同样重要的是, 也需要能够清楚地定义为什么受试动物的某些变化是非不良的。应当指出,毒性研究必须限于少量的定量观察点(剂量或浓度水平),尽管生物响应可以随剂量的变化而连续变化。由于这个原因,这些测试指标的定量结果(例如不良效应和无不良效应水平)以及剂量-响应曲线等, 仅代表对真实生物效应的近似描述。(图1中以图形方式显示)。
本文对目前用于识别、描述、定义和使用有关不良效应的概念和方法进行了梳理。所关注的主要是最终为评估人体健康风险而设计的毒理学研究。这些研究确认(或能够确定)不良效应以及这些不良效应发生或不发生的剂量水平。虽然这个方法适用于单一的研究,但应认识到,在对被测试物相关危害的总体评估中,应根据和平衡所有证据的重要性来评估。通常,这将涉及综合考虑其他研究结果, 如: 对相同药物和类似药物的研究和/或在其他动物物种上的研究等。本文仅限于解释在确定的动物实验系统中观察到的效应,但未讨论实验终点本身及其外推以评估对人体健康危害的相关性,尽管这些内容在评估人体健康风险时显然至关重要。
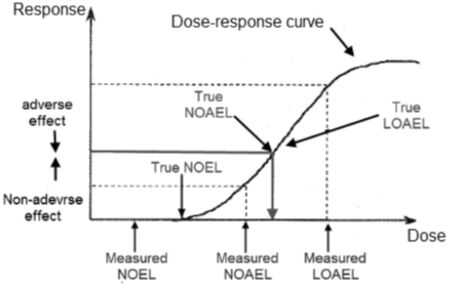
▲图1. 毒性研究的定量结果取决于观察节点
一、
相关定义
本文中描述的用于评估毒理数据的方法使用以下推荐的相关定义。必须注意到,这些定义依赖于毒理数据解释中的最初和关键步骤: 即区分在试药物引起的和自发性变化引起的,并与对照值有差异的这两种不同的变化。
未观察到效应水平(NOEL)
-与适当的对照相比较,在暴露的群体中没有观察到任何效应(不良或非不良)的最高暴露量水平。
未观察到不良效应水平(NOAEL)
-暴露人群与其适当对照之间,不良效应的频率或严重程度没有统计学或生物学意义上的显著性增加的最高暴露水平。在这个水平上可能产生一些影响,但它们不被认为是不良影响或不良影响的前兆。
观察到不良效应最低水平(LOAEL)
-暴露人群与其适当对照之间不良效应的频率或严重程度存在统计学或生物学意义上的显著性增加的最低暴露水平。
不良效应
- 单一或组合的生化,形态或生理变化(对外来刺激的响应),对整个生物体的性能产生不良影响或降低生物体对其他环境挑战作出反应的能力。与不良影响相反,非不良影响可以定义为不会引起影响动物的总体健康,生长,发育或寿命的生化,形态或生理变化的那些生物效应。
生物学上显著的效应
- 生物体或其他生物系统中对生物系统健康具有实质性或显著影响(正面或负面)的响应(对外来刺激)。该概念应该与统计上显著的影响或变化区分开来,这些影响或变化对系统的一般健康状况可能有意义或可能没有意义。
就这些定义达成一致对于制定可用于区分不良和非不良效应的普适性标准至关重要。在危害识别(hazard identification)的,描述无不良效应的最高药物剂量水平的唯一有意义的是未观察到不良效应水平(NOAEL)。然而,未观察到效应水平(NOEL)的概念和使用也渗透到了文献和药物审批的监管过程中。因此,值得注意的是,许多机构实际上使用NOAEL,即使他们可能将其称为NOEL。
显著的生物响应通常有两种类型的。首先,存在正常的生物响应,其表现为对压力的响应,例如运动时出汗,饥饿时减轻体重。这些变化通常代表对外部刺激的正常、稳态反应。其次,存在异常的生物响应,其可能由化学物质或其他压力引起,例如血液恶液质、肝毒性和肾毒性,以及肿瘤。正常的和异常的生物响应都可能在统计学意义上显著地不同于正常的基线值。但后者显然是毒理学家更需要关注的问题。因此,必须谨慎地将统计意义上的差别与真正的不良生物效/响应联系起来。
二、毒理学数据的解读方法
人们普遍认为,评估复杂的多终点毒理学研究的结果并不是一项简单的工作。对毒理学数据进行全面评估将涉及:
• 听取专家意见和判断,需要经验将复杂多样的信息整合到一个的合理的解释中。
• 认识到药物效应可能代表连续性的响应,阈值,或全有或全无响应,等,多种响应类型。
• 在危害特征描述中通常有多种解释的可能。根据证据权重和总体关注程度对结果的描述可能比简单地判定某种效应是否是不良更为恰当和有益。
需要强调的是,关注和评估单个研究可以简化评估过程。主要目的是为确定哪些效应与基线值的差异是与治疗相关,以及其中哪些影响应被视为不良。然而,在实践中,危害评估经常要考虑来自一系列研究的数据(来自不同的物种和研究类型),进行一个综合评估。因此,最终结果反映了所有证据的总体量。下面推荐的评估过程包括2个步骤; 在每个步骤中,列出了在作出判断时应考虑的区别因素。
区别因素A:
用于区分治疗组与对照组之间在治疗相关效应上的随机差异。
如果出现以下情况,观察到的差异不太可能是治疗引起的:
1. 没有明显的剂量-响应关系;
2. 在一个或多个动物中的观察结果可被视为异常值;
3. 被评估终点的检测技术在本质上是不精确的;
4. 在正常的生物变化范围内,即在历史控制值或其他参考值的范围内;
5. 缺乏生物学上的合理性。观察到的差异与同类效应,作用方式或被测试物质的其他已知或预期的效应不一致。
区别因素B:
用于区分不良效应和非不良效应。如果出现以下情况,效应不太可能是不良的:
1. 受试生物体或受影响的器官/组织的一般功能没有改变;
2. 它是一种适应性反应;
3. 它是短暂的;
4. 严重程度有限,低于关注的阈值;
5. 效应是孤立的或独立的。通常未观察到与关注的效应相关的其他参数的变化;
6. 效应不是前兆。这种效应不是已知随着时间的推移而发生的连续变化的一部分,而导致已知的不良效应;
7. 它是其他不良效应引起的;
8. 它与某个特定的实验/动物模型的相关。
限于篇幅,本文将详细地描述并使用实例说明如何使用区别因素A,后续文章详细地描述如何使用区别因素B。这些实例或者是从特定毒理学研究结果中抽取出来的,或者是源自实际经验并为了说明某个问题而构建的有代表性的实例。
三、
确定效应的区别因素A
区别因素A用于区别观察到的差异是否与被测试药物相关。在实践中,可能会采取证据权重方法(即需要考虑各种因素的组合,以便在研究和对照群体之间区分真实效果和其他差异)。对于大多数毒性研究而言,唯一有效的研究内的比较方法是:在研究过程中的每个时间点进行治疗组和同时进行的对照组之间的比较。
然而,当基线数据或其他先前的数据可用时(如在非啮齿类动物的毒性研究中),评估与这些先前值的差异可能是评估观察到的差异是否与治疗相关的最佳比较。这种比较通常适用于短期测试,也适用于长期测试中随时间变化很小或没有变化的指标,例如一些血清化学指标。无论评估何种指标,确定哪些影响是不良效应的方法仍然是相同的。
A-1: 如果没有明显的剂量-响应关系,观察到的差异就不太可能是药物治疗的效应
在常规检测(例如体重,饲料消耗,临床病理学和器官重量)中,当评估低剂量和中剂量组的结果是否有统计学的显著性差异时,使用剂量-响应关系来判断会特别有效。
统计评估变异性大的数据时,经常有机会出现治疗组和对照组之间存在显著差异的情况。在这些情况下,缺乏剂量-响应关系通常是确定该差异与被测试药物治疗无关的充分论据。因此,该差异也不是药物的引起的效应。
然而,有些情况可能无法用剂量-响应关系来评估。如:
• 变化仅限于高剂量组。在这种情况下,如果其他区别因素A不适用,则这些变化可能是药物引起的,是需要评估的。
• 在较低剂量下观察到的终点变化被在更高剂量时的明显毒性(例如,死亡)所掩盖。在这种情况下,不可能评估完整的剂量-响应关系。
• 中剂量或低剂量组的观察到的效应:在一些研究中,如致癌性和神经毒性研究,在中剂量和/或低剂量组中观察到差异可能在高剂量组中不存在。这是由于不同的药物作用机制引起的,这些作用机制是剂量依赖性的(例如,较低剂量时的神经刺激反应和较高剂量时的神经抑制作用)。虽然没有连续的剂量-响应关系,但这种差异应视为药物的效应。
A-2: 如果由于动物个体之间响应的巨大差异可以被认为是异常检测值(Outlier),则该差异不太可能是药物治疗的效应
异常值是极端的个体检测值(高或低),它们与一组数据的主体和历史控制值大相径庭。它们是被认为是不正确的或与其他原因(例如,疾病状态)直接相关的观察和/或检测值。这可以通过独立的观察或其他先前已有的信息来判断。例如,异常值可能是由于未观察到的技术错误,这可以通过目视检查数据,使用散点图或适当的统计方法来判定。仅在包含异常值时才可识别的两组数据之间的差异就不能认为是药物引起的。在某些情况下,异常值可能与特定的临床疾病相关; 这方面的一个例子是具有肝肿瘤的动物,其肝功能数据就会呈现异常。这个异常数据本身并不意味着被测试药物对整个实验动物群体有相关效应。因此,应该忽略该异常值并重新计算均值,以确定潜在的趋势是否仍然存在。
作为另一个例子,理解异常值的影响在生殖和发育毒理研究中很重要。在这些研究中,变化通常取决于产仔数(litter size)。一个例子是胎儿/幼崽的体重。在一定范围内,总窝重(the total litter weight)相对恒定,但在产仔数极端的情况下,几个小的或非常大的胎儿/幼崽可能使平均体重偏离正常值。然而,这与被测试药物无关。在这种情况下,可能有必要评估产仔数的分布,并将大小相似的测试窝和对照窝相比较,以确定观察到的效应是否真实。然而,少数动物可能对药物相关效应更敏感的可能性也不应低估。
A-3: 如果对评估终点的检测方法本身不够精确,则观测到的差异不太可能是药物治疗的效应
评估治疗组和对照组之间的差异应包括对检测方法的固有精度之评估。检测方法的不精确可能源于人类和/或仪器的因素,如可重复性,技术因素和偏差。因此,治疗组和对照组之间的差异可以简单地由于检测/观察的精确度不足而发生。另外,一些数据(例如,接近零为正常值的分位数数据,quantile data with a normal value near zero)的性质使得小的变化在统计上与对照组不同,而它们实际上彼此不可区分。
A-4: 如果差异在正常生物变异范围内 - 在历史控制值或其他参考值的范围内,则不太可能是药物治疗的效应
应将历史控制数据的使用视为更好地理解当前研究中观察到的事件或明显差异的工具。它们不应被视为忽略不想看到的,或难于解释的观测结果的便利工具。同时进行对照组研究的目的是提供一个未治疗动物观测值的正常分布。不可避免地,用于将动物分配给实验组的选择过程和实验/对照组的大小使得平行对照组仅能代表整个对照群体的一个近似值。使用更多的对照数据通常是有益的,因为它可以更准确地揭示对照群体的真实均值和该均值的变异性。
历史对照数据有3种主要使用方式:
1.识别异常对照值
为了识别异常对照值,需要了解平行对照组是否与更大的对照群体一致或是否是非典型的。当确定平行对照组是非典型时,应检查个体动物数据以确定高值或低值是否落在历史范围之外并且是计算平均值时的偏差源。如果排除(或不存在)此类异常值(outliers)后,平行对照组的平均值仍在历史范围的边缘或之外,而治疗组的平均值在历史范围内,则对照组与治疗组之间的差异可以认为是与治疗无关。然而,需要首先考虑历史数据的漂移或当前研究的流程,以解释这种偏差。只有在排除这些可能后,才可以认为对照值超出了正常范围。
表1显示了在大鼠中进行的为期4周的吸入毒理研究报告中的睾丸重量数据。除高浓度组外,所有治疗组的睾丸绝对重量均在统计学意义上显著降低。但没有观察到浓度 - 响应关系,睾丸的相对重量也没有统计学上显著的变化。同时,没有获得与对照组不同的组织病理学数据。此外,治疗组的绝对睾丸重量的平均值在15个其它同样的研究的历史对照值范围内(Mean+SD:3.19 + 0.15g, range: 2.88-3.36g),而平行对照组的平均重量甚至高于该平均值, 3.19g。有鉴于此,平行对照组可以被认为是非典型的,治疗组中睾丸重量的降低可以被认为是异常平行对照值引起的,而不是与药物治疗相关的。

▲表1. 大鼠睾丸的绝对重量数据
2.评估低发生率现象的相关性
对于低发生率的数据,历史范围值对于将药物的真实效应与平行对照组的虚假差异区分开来是非常重要的。使用历史对照数据常常有助于评估低发病率数据(例如,肿瘤的发病率,某些胎儿畸形)。由于低发病率的性质,治疗组可能呈现出低的自发发生率,而对照组的发病率更低甚至完全没有发生。在这些情况下,历史对照数据将提供有关该发现的总体自发发生率的信息。如果治疗组的发生率落在较大的群体值范围内,则可以认为与平行对照组的差异不是药物相关的。
3.了解高发生率结果的相关性
根据定义,高发生率的现象是常见事件(例如,在发育毒性研究中在外部,内脏和骨骼检查中观察到的变异体)。历史数据将呈现被测物种相关数值的正常范围。
表2说明了在新西兰白兔中通过管饲法进行的发育毒性研究的胎儿骨骼相关数据。数据显示所有剂量组中骨骼异常的发生率有增加和减少。在一些情况下,虽然观察到具有统计学意义的差别,但治疗组的发生率明显地落在特定异常的历史范围内。
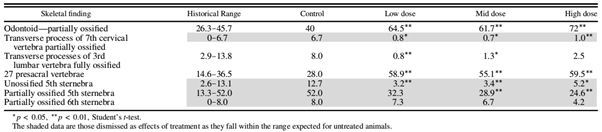
▲
表2. 新西兰白兔的胎儿骨骼异常的发生率(%)
需要着重强调,作为一般原则,需要人为地判断历史数据集是否适合用于在更广泛的自然变异的背景下评估给定研究的结果。历史数据的选择标准已被提出(见参考文献)。选用的最低规范是:毒理研究需要在从同一动物供应商处获得的相同品系,年龄和性别的实验动物上进行;历史数据应来自同一个实验室,并且与被评估研究在实施时间上的差别尽可能地小。在理想情况下,历史对照数据应来自仅包括适当地先于或后于被评估研究的特定时间段内进行的研究;对可能会影响最终结果的研究方法也需确认其一致性:例如,抽样前状态(如禁食或非禁食),研究参数的分析方法,识别病变的组织病理学标准,终端牺牲的时间,等。同时,应当谨慎使用未指明研究方法的正常范围文献值。
A-5: 如果缺乏生物学合理性,观测到的差异就不太可能是药物治疗的效应。
缺乏生物学合理性意味着观测到的差异与同类效应,作用方式或被测试药物已知或预期的效应不一致。
在许多情况下,被测试药物的生物学,化学或物理学性质,和/或相同或类似药物的测试结果有助于确定对被测试药物的响应是否在生物学意义上合理。例如,在某些情况下,如果被测试药物引起的效应明显超出其生物学/生理学的效应范围,则可以排除相关检测结果是该药物引起的可能。这些检测量结果最可能来自实验错误或人为错误。实例包括体重高于物种/品系可达到的最大值,负饲料消耗值和生理学上不可能的血液学/临床化学测试值。
总结和前瞻
在评估药物的内在毒性时,有两个关键因素:(1)有关任何一种不良反应的性质和重要性的知识;(2)未观察到或首次观察到这种效应的剂量或暴露水平。为了获得这些信息,需要对生物系统中的不良效应的概念有一致的定义和应用。一个评估毒理学数据的通用方法对于实现毒理数据解释的一致性至关重要。
大量研究和实践经验表明一个两个步骤的评估过程,或者说两个决策层面,是最适用和合乎逻辑的。首先,需要区分那些治疗组中观察到的效应与对照组之间的差异是否是被测试药物引起的。只有在确定了那些真实的,与药物相关的效应之后,便可以在第二步中进一步评估这些效应,以区分哪些不良效应,哪些是非不良效应。
每个决策层面都确定了一组区别因素,A和B。应用这个评估流程来解释毒理数据可以得到更一致的结果。限于篇幅,本文仅详细地描述并使用实例说明如何使用区别因素A,包括如下问题: 1. 测试结果有没有明显的剂量-响应关系; 2. 在一个或多个动物中的观察到的结果是否可被视为异常值; 3. 被评估终点的检测技术在本质上是否是不精确的; 4. 测试结果是否在正常的生物变化范围内,即在历史控制值或其他参考值的范围内; 5. 测试结果是否缺乏生物学上的合理性,即观察到的差异是否与同类效应,作用方式或被测试物质的其他已知或预期的效应不一致。后续文章详细地描述如何使用区别因素B,敬请关注。
总而言之,评估复杂的多终点毒理学数据并不是一项简单的工作。全面地评估毒理学数据需要专家的意见和判断。在实践中,重要的是要采取证据权重平衡的方法,即通常需要考虑已识别的各种影响因素的组合,已有数据的相对重要性和可靠性,以解释单一毒理学研究的数据或某个被测试药物的所有毒理学数据。
特别声明
本文如有疏漏和误读相关指南和数据的地方,请读者评论和指正。所有引用的原始信息和资料均来自已经发表学术期刊、官方网络报道等公开渠道,不涉及任何保密信息。参考文献的选择考虑到多样化但也不可能完备。欢迎读者提供有价值的文献及其评估。
扩展阅读
1.
如何决定单克隆抗体药物在首次人体临床研究时的起始剂量(一):NOAEL方法
参考文献
1. US Food Drug Administration. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. Rockville, MD: Food and Drug Administration; 2005. [Accessed July6, 2005]. Available from: http://www.fda.gov/downloads/drugs/guidances/ucm078932.pdf.
2. Suh HY,et al, Determination of the starting dosein the first-in-human clinical trials with monoclonal antibodies: a systematic review of papers published between 1990 and 2013 . Drug Des Devel Ther. 2016;10: 4005–4016
3. LewisRW, et al (2002) Recognition of Adverse and Nonadverse Effects in Toxicity Studies T OXICOLOGIC PATHOLOGY , vol 30, no 1, pp 66–74, 2002
4. AshbyJ, Lefevre PA (2000). The prepubertal male rat assay as an alternative to the Hershberger castrated male rat assay for the detection of antiandrogens,oestrogens and metabolic modulators. J Appl Toxicol 20: 35–47.
5. Burger GT, Renne RA, SagartzJW, Ayres PH, Coggins CRE, Mosberg AT, Hayes AW (1989). Histologic changes in the respiratory tract induced by inhalation of xenobiotics: Physiologic adaptationor toxicity? Toxicol Appl Pharmacol 101: 521–542.
6. Chang PK, O’Hara GP, Hayes WA(1982). Principles and methods of acute and subchronic toxicity. In: Principles and Methods of Toxicology, HayesWA (ed). Raven Press, New York, pp 1–52.
7. EPA (1995). The Use of the Benchmark Dose Approach in Health Risk Assessment. EPA/630/R-94/007.
8. Hamada C, Yoshino K, Abe I,Matsumoto K, Nomura M, Yoshimura I (1998). Detection of an outlier and evaluation of its influence in chronic toxicity studies. Drug Infor J32:201–212.
9. Haseman JK, Huff J, Boorman GA(1984). Use of historical control data in carcinogenicity studies in rodent.Toxicol Pathol 12: 126–135.
10. Miller FJ (1995). Uptake and fate of ozone in the respiratory tract. Toxicol Lett 82/83: 277–285.
11. Morgan KT (1997). A brief review of formaldehyde carcinogenesis in relation to rat nasal pathology and human health risk assessment . Toxicol Pathol 25: 291–307.
12. Paynter OE (1984). Oncogenic Potential:Guidance for Analysis and Evaluation of Long Term Rodent Studies. Evaluation procedure #1000.1 Office of Pesticide and Toxic Substances, EPA, Washington,DC.
13. Willard MD, Twedt DC (1994).Gastrointestinal, pancreatic, and hepatic disorders. In: Small Animal Clinical Diagnosis by Laboratory Methods. 2nd ed, Willard MD, Tvedten H, Turnwald GH(eds).WB Saunders Pub.
欢迎加入小编团队成为小编一员










