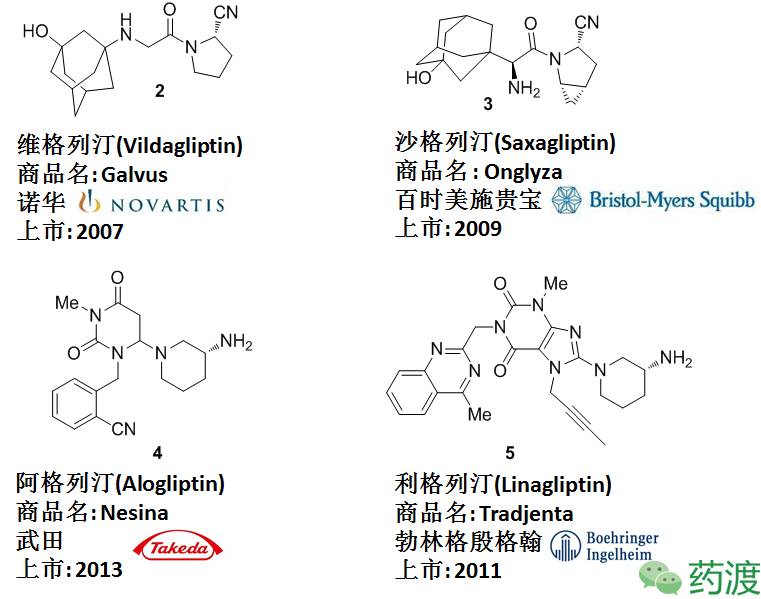
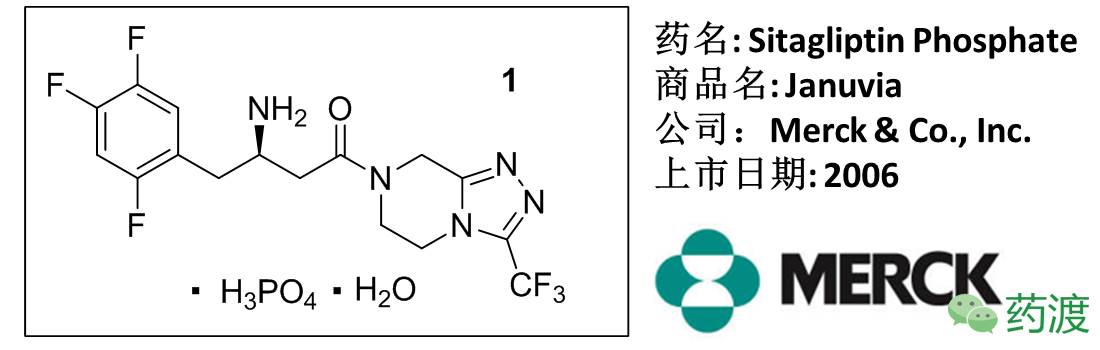
西他列汀(捷诺维)——首例FDA批准(2006)上市的DPP-4抑制剂,用于治疗2型糖尿病。临床显示,西他列汀有很高的安全性和稳定性,其他同靶点药物如下图。作为2016年全球销售排名前十的药物(64.4亿美元),其合成路线的进化过程也显示了如今化学工业正在努力寻找生产成本低廉,操作简便,清洁环保之间的最优平衡点。
西他列汀的合成工艺大致可分为三代,第一代使用具有手性中心的原料,通过保护基团策略,经多步合成产物,但使用保护基会使原子经济性下降,以及冗长的合成步骤是第一代路线主要的不足。第二代使用手性助剂构建手性中心,依然无法摆脱当量化学废料的生成。第三代直接使用不对称催化氢化的方法构建手性中心,且不需要使用保护基团,不但简化了合成步骤,还降低了消耗,提高了原子经济性。
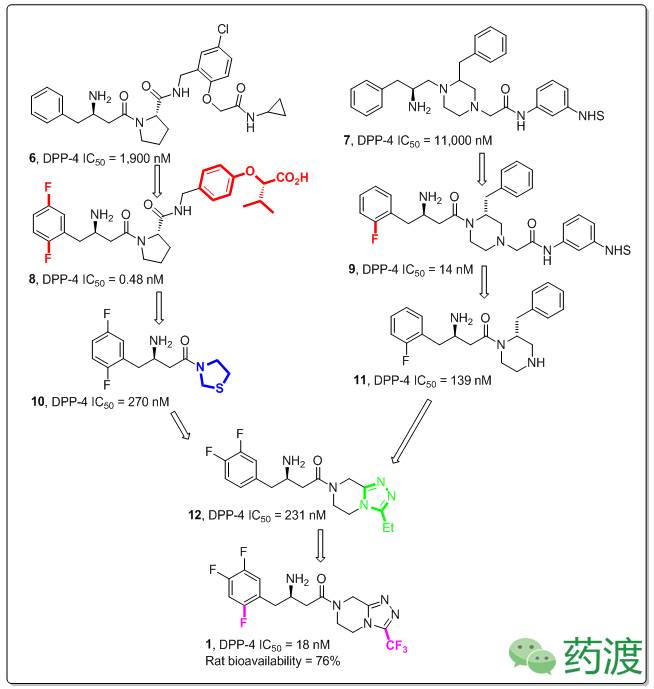
西他列汀最初是以脯氨酸(
6
)和哌嗪(
7
)为母核,通过在芳环上引入F原子和更换苄胺苯环上的基团使IC50有明显改变,得到
8
和
9
化合物,但是两个化合物的口服生物活性太低(F
rat
< 1%),进一步简化化合物结构得到分子
10
,
11
,这两个分子又因为噻唑烷和哌嗪环在生物体内代谢不稳定导致生物活性很低而不能成药,更换三氮唑基团后提高了分子体内代谢稳定性,得到分子
12
,最终将乙基更换为三氟甲基,在芳环临位再引入一个F原子得到药物分子
1
,综合考虑效价、靶点选择性和临床前药动学之后确定的最优分子结构为
1
。
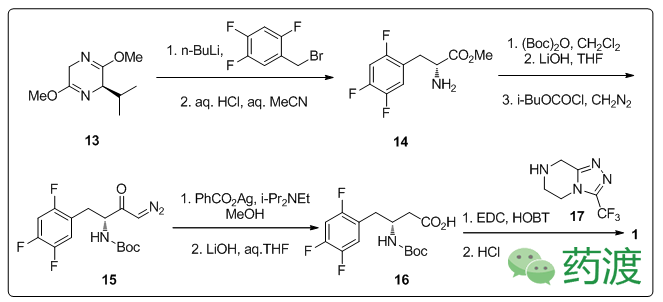
第一代合成方法,分子
1
的合成过程由
13
开始,经Schöllkopf's bis-lactim合成法,亲核取代,酸性条件下水解得到α-氨基酸
14
,之后Boc保护,重氮化,经Arndt-Eistert得到延长碳链的β-氨基酸
16
,与
17
反应得到分子
1
。
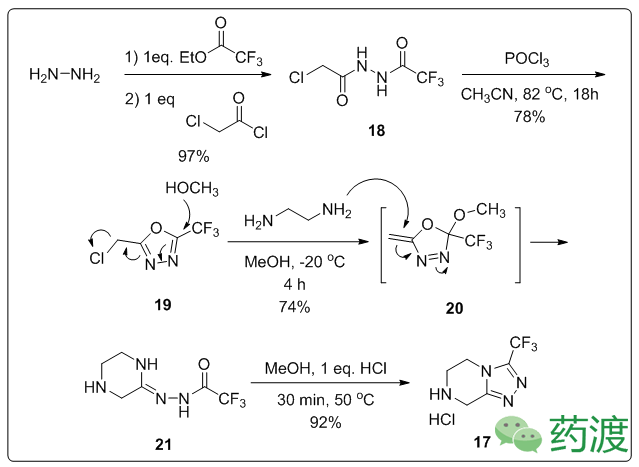
三氮唑分子
17
的合成,先由三氟乙酸乙酯与水合肼缩合,再与氯乙酰氯发生取代反应得到分子
18
,POCl
3
作用下脱水环化得到分子
19
,甲醇溶液体系中加入乙二胺得到分子
21
,最后甲醇溶液中加入盐酸可得
17
,产物以盐的形式直接析出,该方法的工业化总产率为52%。
手性中心还可以通过不对称氢化的方式构建——
23
合成手性分子
24
。羧酸分子
22
在Masamune反应条件下转化为
23
,以(S) -Binap RuCl为手性催化
剂 (< 0.1 mol%)催化氢化,83%的产率得到
24
,并且有94% ee,经Mitsunobu反应得到内酰胺分子
25
,在碱性条件下水解得到分子
26
,之后与分子
17
反应得到
27
,钯碳氢气条件下除去保护基团,酸化得到磷酸盐分子
1
。这一方法虽然可以大规模工业化生产分子
1
,但是分子
25
使用苄氧基作为保护基团使整个过程原子经济性下降,同时产生大量化学废料。
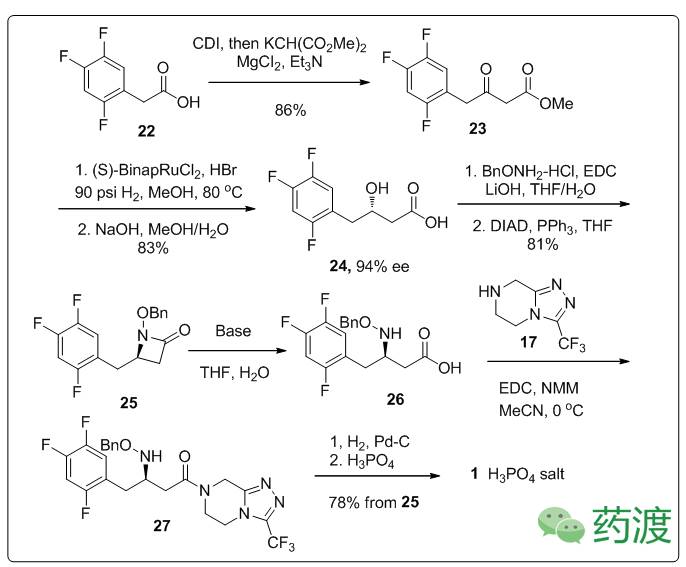
为了能够突破这一局限,默沙东的研究团队决定对还原步骤进行优化。使用(S)-苯甘氨酰胺手性助剂((S)-phenylglycinamide chiral auxiliary (PGA)),由
23
直接得到
28
,之后对还原体系的筛选发现,PtO
2
在HOAc体系下还原效果最好,可以以90%收率,91%的过量非对映异构体得到目标产物
29
,之后与分子
17
反应,脱手性助剂就得到分子
1
。
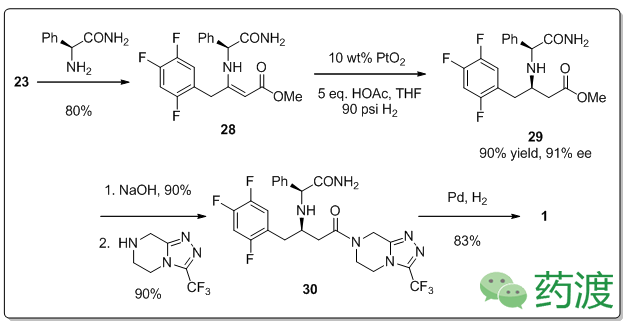
对比默沙东第一代合成方法,使用PGA合成中间体可以大大减少反应步骤数。之后,在合成初期就将三氮唑部分连接可以进一步减少合成步骤。分子
31
与手性助剂合成分子
32
,在PtO
2
,THF/MeOH条件下还原得到分子
33
(93% yield,97.4% de),最后西他列汀在L-酒石酸中分离,PGA在重结晶过程中除去。
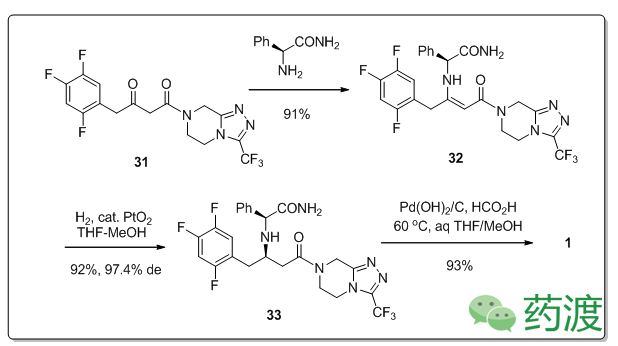
尽管使用(S) -PGA手性助剂可以将整个合成步骤缩短,但是依然会产生副产物2-苯乙酰胺。而为了实现大量工业化生产,就不能使用手性助剂来构建产物的手性中心,因此,研究者将研究重点放在了中间体
37
上。
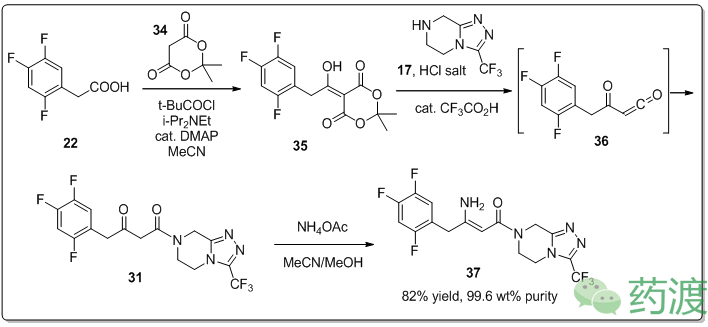
分子
22
被活化之后与
34
反应得到
35
,在经历烯酮中间体
36
,捕获分子
17
之后生成分子
31
,再与乙酸铵反应得到分子
37
,总产率82%,后处理仅需简单抽滤即可得到99.6 wt%产物。而最后一步就是对氨基未保护分子
37
的不对称氢化。
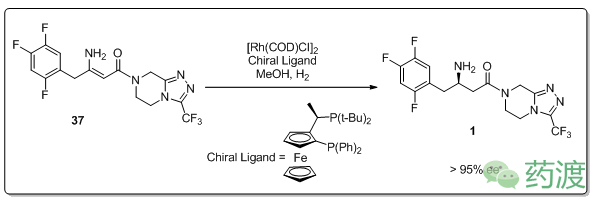
筛选常用的氢化金属钌(Ru)、铑(Rh)、铱(Ir),并结合不同的催化体系,最终确定了最优反应催化剂是[Rh (COD) Cl]
2
,而手性配体方面,通过综合考虑产率,ee值和成本后,确定以t-Bu-JOSIPHOS为最优配体。
向体系内加入少量三氯化铝(0.15-0.3 mol%)可以提高反应的对映选择性和转化率,但是机理尚不清楚。该步骤99.6 wt %,大于95% ee值,体系压力提升到250 psi时,可以将催化剂用量降低到0.15 mol%,反应后通过使用Ecosorb C-941可以将90-95%的金属催化剂回收,重结晶以79%的产率得到西他列汀(> 99% ee,> 99%» purity)。
通过使用不对称氢化方法,很好的规避了氨基保护基团的残余问题,并很好的实现了手性中心的构建,相对于默沙东第一,二代西他列汀合成方法,该合成路线更加环保,步骤更少,原子经济性更高,每公斤西他列汀成品合成所产生的废料由250公斤降低到50公斤,而整个过程对水的消费量为零。
该方法也因在环保方面的贡献而获得了2006年总统绿色化学奖(Presidential Green Chemistry Award),2005年IChemE AstraZenecaAward,以及2009年Thomas Alva Edison Patent Award。
参考:
1. Prix Galien USA Award (2007) and the Thomas Alva Edison Patent Award (2007) for Merck's U.S. Pat. 6,699,871.
2. Expert Opin. Investig. Drugs. 2008,77,1559-1565.
3. Eur. J. Pharm. 2009,623,148-154.
4. Bioorg. Med. Chem. Lett. 2004,14,5151-5155.
5. Bioorg. Med. Chem. Lett. 2004,14,4763-4766.
6. J. Med. Chem. 2005,48,141-151.
7. J. Org. Chem. 1962,27,3243-3248.
8. Org. Process Res. Dev. 2005,9,634-639.
9. Org. Lett. 2005,7,1039-1042.
10. J. Am. Chem. Soc. 2004,126,9918-9919.
11. Org. Process Res. Dev. 2006,10,723-726.