验证的定义
2010版GMP对确认和验证的定义是:
验证:证明任何操作规程(或方法)、生产工艺或系统能够达到预期结果的有文件证明的一系列活动。
1. 有文件记录
2. 有预期的结果
3. 一种证明活动,而非开发。
确认与验证的范围
设备设施
◦ 制造、包装设备,纯水、空调、压缩空气系统
工艺
◦ 每个产品制造、包装工艺
清洗规程
◦ 与产品直接接触设备的清洗流程-手工&自动
分析仪器和分析方法
◦ 天平、液相色谱、崩解仪,原辅料、成品分析方法
计算机系统
◦ SAP系统,EMS系统,MES系统
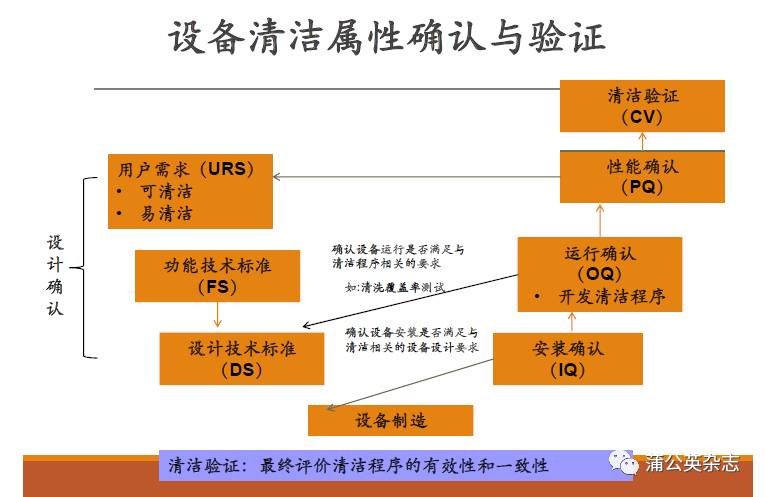
通用的验证框架体系
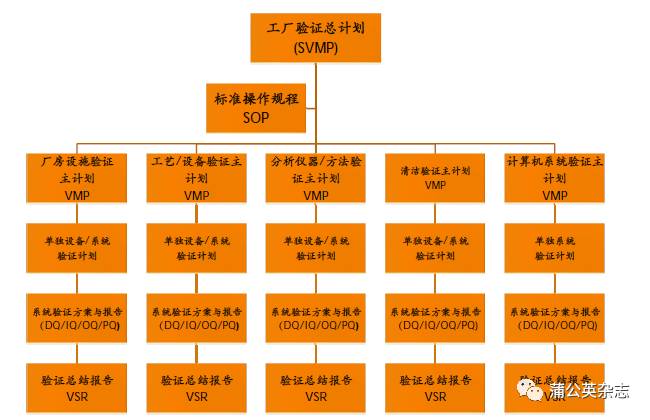
验证总计划
定义:
验证总计划是提供整个工厂验证行为概况的文件。
验证总计划是验证文件框架中最高级别文件,概述了公司概况,验证的指导方针与策略(验证方法,变更控制,偏差处理等),公司各个部门或职位在验证活动中的职责,公司内部涉及的验
证活动计划与状态等。
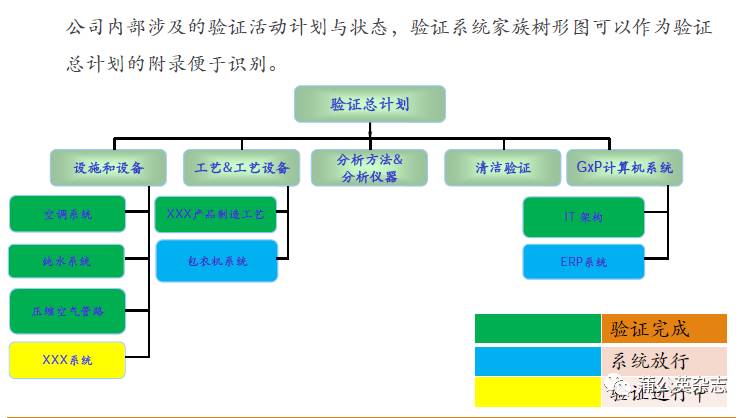
产品为主线的验证总计划
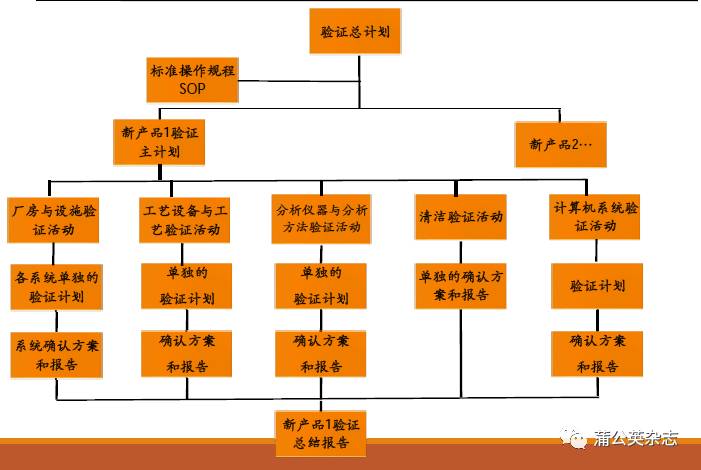
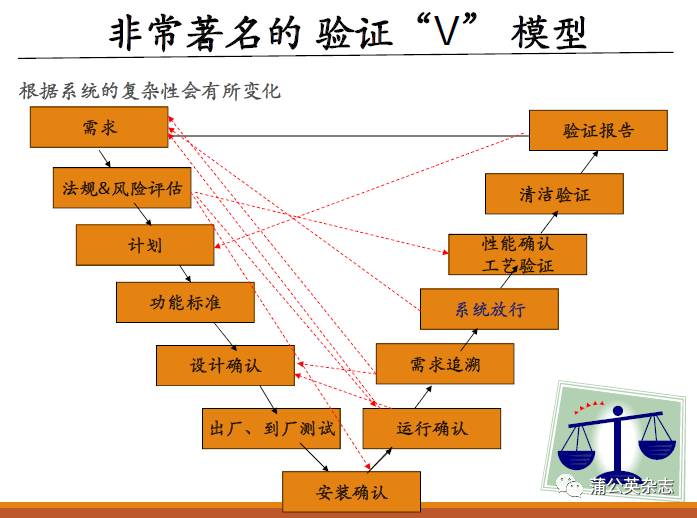
验证是什么样的过程?
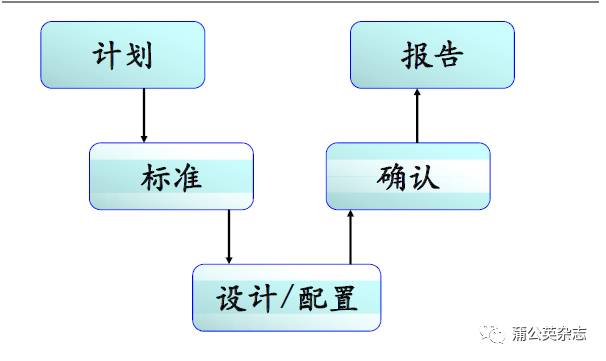
验证的实施步骤
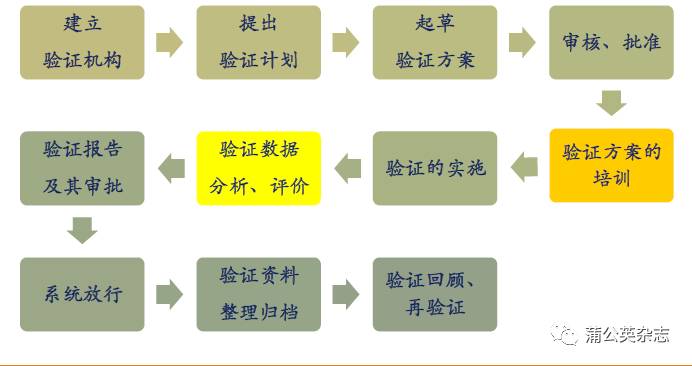
验证生命周期文件
验证的一个基本特性是其工作是按照事先批准的方案进行的。因此验证方案为确定在验证过程中采取的行为进行规定的关键文件。
每个验证工作都必须准备验证方案并在验证实施前获得批准。
验证方案的接受标准
对于每个系统的验证文件以及接受标准应该在验证计划中进行详细的规定。
对于验证方案中的每一个测试都应该有与之相对应的唯一接受标准。
记录所有相对于方案的偏差及不符合接受标准的不符合事件,对这些事件应进行调查、说明并存档。
验证报告
1. 在验证行为的每个确认步骤完成以后及在整个验证行为完成以后,应该对验证行为进行回顾以确保所有的数据都被适当地记录与报告。
2. 最终的结论必须被认可并得到批准并清楚地表明该接受还是拒绝验证下的项目。
3. 该报告应包括所有失败的测试及随后的整改行为。
清洗验证的定义
有文件和记录证明所批准的清洁规程能有效地清洁设备,使之符合药品生产要求.
2010年版GMP正文:
第一百四十三条清洁方法应当经过验证,证实其清洁的效果,以有效防止污染和交叉污染。
清洁验证应当综合考虑设备使用情况、所使用的清洁剂和消毒剂、取样方法和位置以及相应的取样回收率、残留物的性质和限度、残留物检验方法的灵敏度等因素。(2010年版GMP正文)
清洗验证的法规与指南
FDA(cGMP):21CFR-PARTS 210 & 211
211.67设备清洁与维护
间隔一定时间应该对设备和用具进行清洗、维护和消毒,防止可能的故障和污染及改变药品安全性、均一性、效价、质量和纯度。
EU GMP:
336 生产设备的设计必须容易进行彻底清洁,必须按照详细的书面操作规程进行清洁并储存在清洁干燥的条件下。
337 洗涤和清洁设备必须正确选择,保证在使用时不会带来污染。
WHO-937-Appendix 3 : Cleaning validation
1.4 清洁验证目的在于证明设备始终符合产品、清洗剂和微生物残留验收要求,以预防可能的污染和交叉污染。
指南:ICH Q7
FDA《清洁验证检查指南》
Ø批准/受控的清洗程序
Ø验证方案
Ø经过验证的化学/微生物检测方法
Ø批准的验证报告(结论:清洁程序有效,达到可接受标准)
Ø有效的培训/变更控制
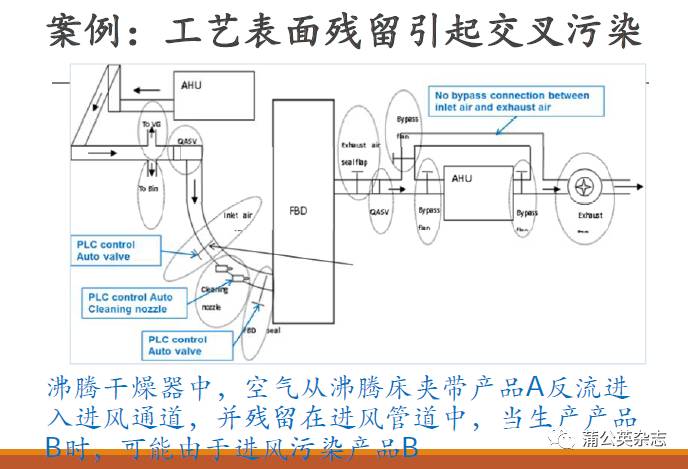
清洗验证的要求
GMP 的要求以防止交叉污染
良好的商业习惯
法规部门的要求

清洗验证的前提条件
健康& 安全
- 对于所有的产品及清洗剂是否具有相应的健康与安全数据?
- 是否对清洗操作进行了评估
设备& 设施的设计
- 设计是否易于清洗与检查
- 是否对自动系统进行了确认(e.g. 在线清洗系统)
清洗规程的开发
- 清洗规程是否全面,一致,详尽,准确并且经过批准
- 是否定期对清洗规程进行回顾?
最初的评估阶段
应尽早准备清洗验证主计划并批准
必须确定清洗验证的范围与目的并书面进行记录
考虑所需要进行清洗验证的水平
开始最初的数据收集
设备的评估
将设备/部件按下述情况进行分类:
接触产品- 应该在清洗验证中进行确认
不接触产品- 一般无需在清洗验证中进行确认
偶尔接触产品– 应该在清洗验证确认该类设备/部件为“目测洁净,干燥并且无嗅”.
关于是否某个产品/部件是否包含在清洗验证中应该在理论依据的章节进行书面的记录。
将产品接触及偶尔接触产品的设备/部件按下述方式分类:
◦ 一次性使用, 一次性使用的部件(e.g. 小桶的衬垫, 某些过滤器)
◦ 某个产品专用部件(e.g. 压片机的冲子,震荡筛的筛网)
◦ 多个产品公用的部件
每个部件的分类必须注明原因并进行详细的记录

用户需求
合规需求
◦GMP中关于设备清洁的要求
工艺需求
◦清洁工艺需求(目标产品的清洁需求)
管理需求
◦控制管理需求
GMP的需求
第七十一条设备的设计、选型、安装、改造和维护必须符合预定用途,应当尽可能降低产生污染、交叉污染、混淆和差错的风险,便于操作、清洁、维护,以及必要时进行的消毒或灭菌。(正文)
第七十四条生产设备不得对药品质量产生任何不利影响。与药品直接接触的生产设备表面应当平整、光洁、易清洗或消毒、耐腐蚀,不得与药品发生化学反应、吸附药品或向药品中释放物质。(正文)
第七十六条应当选择适当的清洗、清洁设备,并防止这类设备成为污染源(正文)
第四十二条验证应当考虑清洁方法的自动化程度。当采用自动化清洁方法时,应当对所用清洁设备设定的正常操作范围进行验证;当使用人工清洁程序时,应当评估影响清洁效果的各种因素,如操作人员、清洁规程详细程度(如淋洗时间等),对于人工操作而言,如果明确了可变因素,在清洁验证过程中应当考虑相应的最差条件。(附录)
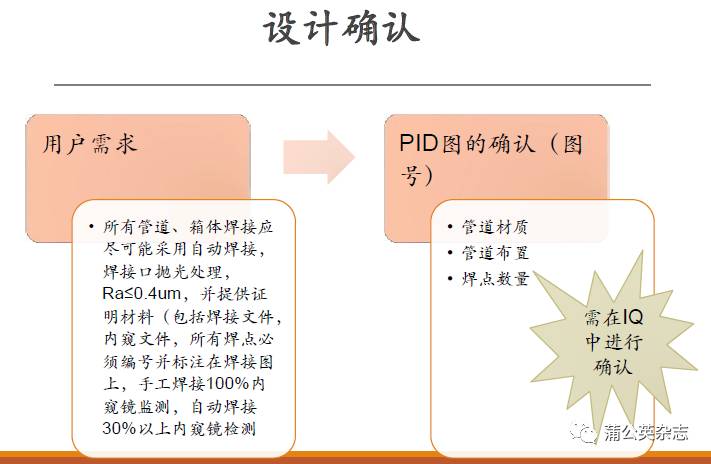
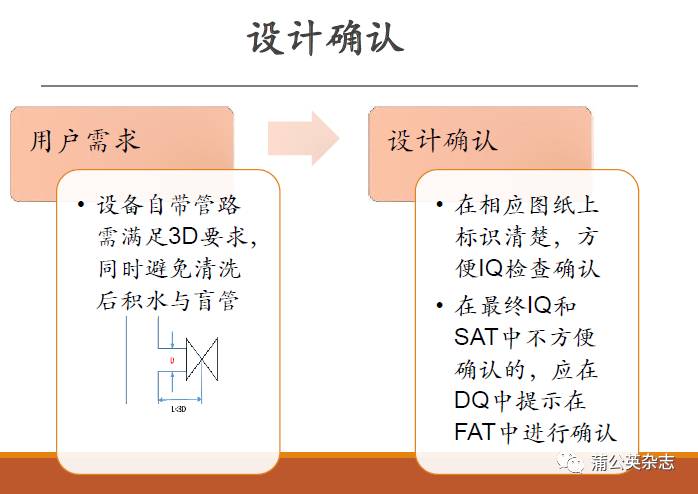
具体化用户需求-总体要求
设备有在线清洁功能,应确保冻干箱和冷阱所有地方都能清洗到,应确保无死角,清洗能覆盖箱体内表面
◦板层可实现动态清洗,在机房与无菌室内均设有控制升降按钮
◦视镜内部须有清洗头,以保证清洗冻干机时能有效对其进行清洗


具体化用户需求-附属设施
设置外置清洗站,配置单罐双泵系统,储罐能满足CIP要求
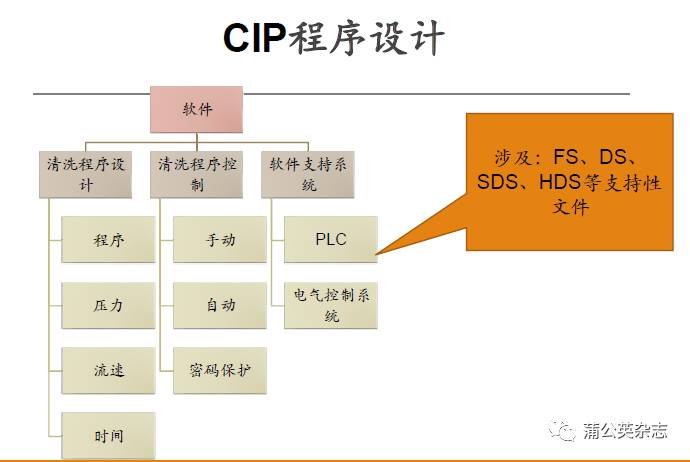
具体化用户需求-清洗程序
1. 可调用固定的清洗程序,自动清洗冻干箱、冷阱及相关部件,清洗程序通过三级密码权限保护
2. 操作方便:手动和自动,可切换
3. 清洗管道及结构设计必须考虑对冻干箱和冷阱分步清洗
4. 冻干小门在执行清洗与灭菌等程序前必须具有自锁功能


设计需求
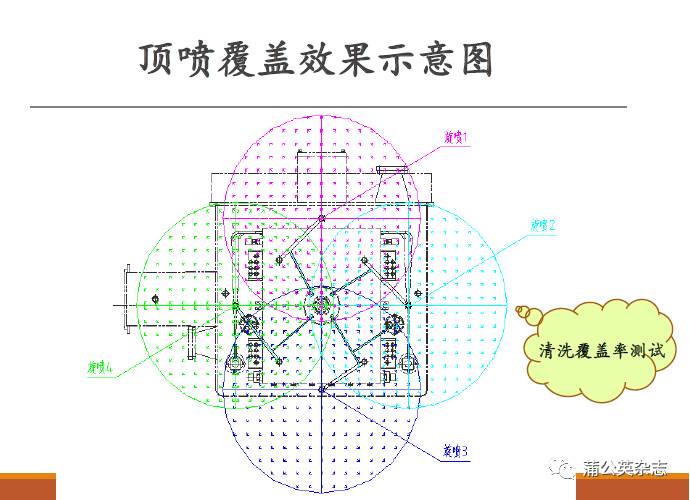
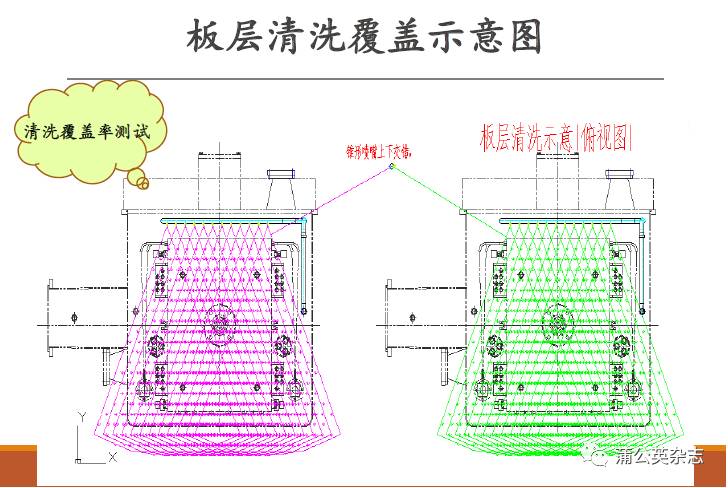
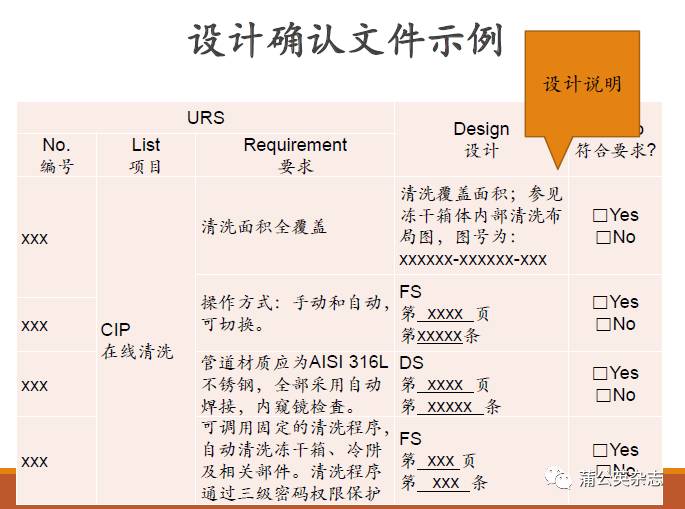
URS:在线清洁,对清洗喷头必须做喷淋覆盖测试,清洗面积全覆盖
首先明确需要清洗覆盖的部位
◦箱体内表面
◦板层外表面
◦观察窗视镜
◦冻干箱门
◦…..

产品的评估
对所有相关的产品获得如下的信息:
◦ 活性成分
◦ 批量
◦ 标准日剂量
◦ 毒性
◦ 溶解性
◦ 生产量
◦ 暴露剂量
产品的辅料是否对清洗规程产生不利的影响?
◦ 阻止设备表面变湿
◦ 粘在设备表面
◦ 颜色,香味或其他气味
◦ 不溶解
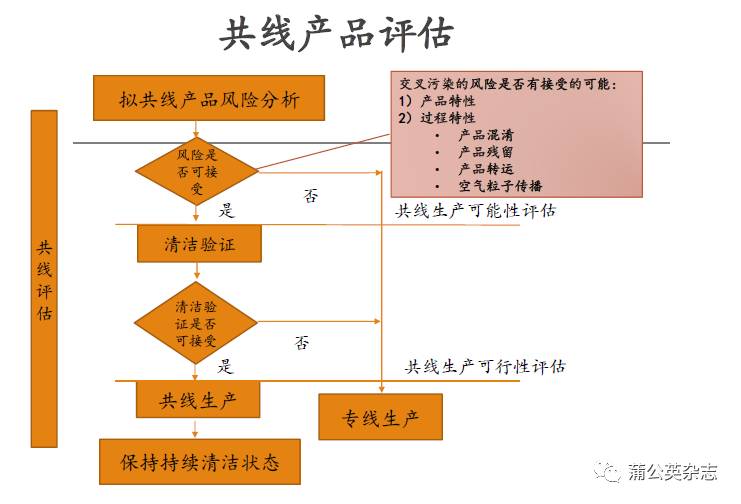
共线产品的评估
法规要求:
第四十六条为降低污染和交叉污染的风险,厂房、生产设施和设备应当根据所生产药品的特性、工艺流程及相应洁净度级别要求合理设计、布局和使用,并符合下列要求:(2010年版GMP正文)
(一)应当综合考虑药品的特性、工艺和预定用途等因素,确定厂房、生产设施和设备多产品共用的可行性,并有相应评估报告


清洗规程的评估
◦ 是否包括了所有的设备/清洗操作?
◦ 清洗操作是手动还是自动的(是否具有在线清洗系统)?
◦ 清洗规程是否足够详细以便可以进行持续一致的操作?
◦ 是否规定了干燥的方法以及干燥方法是否正确?
◦ 标准操作规程是否规定了需要检查与记录的信息?
◦ 清洗规程是否包括了状态标识的要求?
◦ 是否详细描述了在完成检查后如何确保存储是安全的?
清洗规程的重要信息
◦设备拆卸要求
◦所用清洁剂的浓度和体积;或者仅用热水
◦浸泡/搅拌时间
◦冲洗时间和体积
◦清洗剂或者水与产品的兼容性
◦清洗剂/水的温度或设备的清洗温度
◦是否加压或者使用机械手段
◦建立并控制时间节点:
◦ 在设备使用后到开始清洗的时间间隔
◦ 在各个清洗步骤之间的时间间隔
◦设备干燥的方法
◦洗液处置
清洗规程的评估
法规规定:
第四十一条验证应当考虑清洁方法的自动化程度。当采用自动化清洁方法时,应当对所用清洁设备设定的正常操作范围进行验证;当使用人工清洁程序时,应当评估影响清洁效果的各种因素,如操作人员、清洁规程详细程度(如淋洗时间等),对于人工操作而言,如果明确了可变因素,在清洁验证过程中应当考虑相应的最差条件。
清洗剂的使用
如果有任何可能–不要使用清洗剂!!
由于需要对清洗剂的残留进行验证,因此清洗剂的使用引入了复杂及潜在的问题
需要完全的信息(e.g. 健康与安全数据以及分析方法) 以确保可以完成清洗剂残留的验证
在某些情况下,清洗剂测定的分析方法验证需要非常专业的知识与人力物力
法规规定:
第四十二条如使用清洁剂,其去除方法及残留量应当进行确认。
清洗验证的矩阵设计
适用于特定的设施与设备
可能会减少清洗验证的工作
进行矩阵设计需要具有公用的设备及共用的清洗方法
最差条件/标记产品的选择必须在“依据”章节中进行解释与判定
标记产品/最差条件
法规规定:
第四十五条当采用最差条件产品的方法进行清洁验证模式时,应当对最差条件产品的选择依据进行评价,当生产线引入新产品时,需再次进行评价。如多用途设备没有单一的最差条件产品时,最差条件的确定应当考虑产品毒性、允许日接触剂量和溶解度等。每个使用的清洁方法都应当进行最差条件验证。
在同一个工艺步骤中,使用多台同型设备生产,企业可在评估后选择有代表性的设备进行清洁验证。


……未……完……
ouryao-com·因为有你










