吉非替尼包合物的制备、表征及体内外考察
Preparation, Characterization and in vitro-in vivo Evaluation of
Inclusion Complexes of Gefitinib
张理星,杜丽平,冯 中,郝贵周
(鲁南制药集团股份有限公司国家手性制药工程技术研究中心,山东临沂 276006)
摘要:
为提高吉非替尼(1) 口服吸收生物利用度,通过机械化学技术,即采用GMS 10-8 型滚动式球磨机分别制备了以羟
丙基-β- 环糊精(HP-β-CD) 与阿拉伯半乳聚糖(AG) 为载体的1 包合物,并采用核磁共振弛豫时间(T2)、扫描电镜(SEM)、差示热量扫描(DSC)、粉末X- 射线衍射(P-XRD) 对包合物进行物理表征。体外试验显示:1/HP-β-CD 包合物将1 溶解度由0.77 g/L 增至2.16 g/L,药物在120 min 时的溶出率由60%增至92%;1/AG 包合物将1 溶解度增至1.32 g/L,药物在120 min 时的溶出率增至75%。大鼠体内试验显示:1 原料药、1/AG 包合物及1/HP-β-CD 包合物的绝对生物利用度分别为56.94%、81.63%和78.48%。本试验制备的1 与AG、HP-β-CD 的球磨产物均为包合物,它们成功地改善了原料药的溶解度、体外溶出及体内吸收行为,可为后续开发低药物剂量、高口服吸收生物利用度的1 剂型提供帮助。
关键词:
机械化学技术;包合物;表征;分子间作用力;溶解度;溶出度;口服生物利用度
吉非替尼(gefitinib,1) 是一种用于治疗接受过
化学治疗或不适合化学治疗的局部晚期或转移性非小细胞肺癌患者的抗受体分子。它可竞争性地结合细胞表面表皮生长因子受体酪氨酸激酶催化区域上的Mg-ATP 结合位点,抑制酪氨酸激酶的磷酸化,阻断下游的信号传导,进而发挥靶向抗肿瘤作用[1—4]。1 因高特异性、高疗效及较好的耐受性在临床上备受瞩目。但已上市的1 剂型为片剂,口服吸收缓慢,生物利用度仅59%,且不良反应程度随给药剂量增加而加重[5]。1 在生物药剂学分类系统(BCS) 中属于Ⅱ类,较低的水溶性是限制其体内吸收的关键步骤[ 6]。因此,本课题组考虑将1 制成包合物,在不影响该药耐受性的前提下,改善1 溶解度及溶出行为,进而提高其口服吸收生物利用度,降低市售片剂中1 的剂量及其剂量依赖性的不良反应( 如腹泻、呕吐)[5,7]。
包合物的传统制备技术主要包括溶剂蒸发法、
捏合法、冷冻或喷雾干燥法与饱和水溶液共沉淀法,但这些技术在制备过程中普遍存在大量的溶剂消耗、原料药被部分降解或聚合等现象[ 8—9]。机械化学技术是通过机械力的不同作用方式( 研磨、压缩、剪切、摩擦、延伸、弯曲、冲击等),引入机械能量的累积,从而使受力物体的物理化学性质发生变化的一门技术[10]。机械化学技术因在制备过程中避免使用有机溶剂、对环境友好无破坏作用、并且一步操作即可制得目标产物等诸多优势,在药剂学领域日益受到关注。现有的机械化学技术主要通过球磨机实现。在球磨过程中,机械能通过碰撞接触点转移到物料表面并被限制在很小的空间内,导致物料表面的能量聚集,形成一个亚稳定区;该区域
必须释放大量的额外能量,物料才能够处于热力学
稳态,该过程直接导致的结果是物料之间发生相转移或化学反应,或是分子间作用力的生成[11—12]。机械化学技术现已作为一种实验手段用于设计、制备原料药的超分子复合物,改善药物的物理化学性质。
本试验采用机械化学技术制备1 包合物,旨
在改善1 的溶解度及溶出行为。阿拉伯半乳聚糖(AG) 因具有较好的水溶性和血管通透性,将其作为1 的枝杈状包合物载体[13—14]。羟丙基-β- 环糊精(HP-β-CD) 因可抑制肠膜上的P- 糖蛋白活性,进而促进药物的吸收,亦被选为1 的空桶状包合物载体[ 9,15]。试验制得的球磨产物采用核磁共振弛豫时间(T2)、扫描电镜(SEM)、差示热量扫描(DSC)、
X- 射线粉末衍射(P-XRD) 进行表征。此外,本课
题组还进一步研究了球磨产物的体内外释药行为。
1 仪器与试药
2 方法与结果
2.1 1 包合物的制备
2.1.1 1/AG 包合物
采用滚动式球磨机[ 球磨罐的体积为300 ml,
球磨介质为不锈钢球( 直径22 mm, 总质量为675.0 g),球磨介质的加速度为1×g],将1 原料药2.00 g 和AG 20.00 g( 摩尔比为14 ∶ 5) 混合,球磨总时间为16 h, 分别在球磨第1、2、4、8 和16 h 取样检测。
2.1.2 1/HP-β-CD 包合物
采用滚动式球磨机同上操作,将1 5.09 g 和
HP-β-CD 16.91 g( 摩尔比为1 ∶ 1) 混合,球磨总时间为16 h,分别在球磨第1、2、4、8 和16 h 取样检测。
2.2 1 包合物的表征
2.2.1 粒度分布
用激光粒度分析仪测定球磨产物的粒度分布。
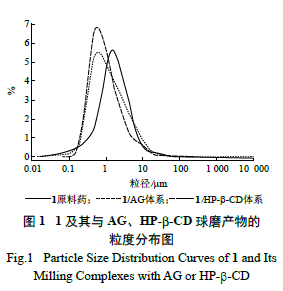
采用马尔文MS3000 Aero S 模块干法测定,参数设定如下:折射率1.62、吸收率0.01、样品测量时间12 s、遮光度0.5 ~ 6、气压250 kPa、进样速度25%、粒径检测范围0.010 ~ 10 000.000 μm。1 及其球磨16 h 产物的粒度分布见图1。3 个体系的微粉粒子分布都较均匀,接近于正态分布。未经机械球磨处理的1,中位粒径为1.54 μm;机械球磨处理的1/AG 体系中,中位粒径降至0.60 μm;机械球磨处理的1/HP-β-CD 体系中,中位粒径降至0.58 μm。上述结果显示,1 与包合载体材料的物理混合体系经机械球磨处理后,体系中原有微粉颗粒的粒径均下降,降幅约为60%。
2.2.2 扫描电镜表征
用扫描电镜观测球磨16 h 产物的外观形貌。首
先,将导电胶带黏附在样品座上,再将待测试的固体样品均匀地撒在上面,用洗耳球吹去未粘住的样品粉末;然后在样品表面镀上一层导电膜( 金粉);最后将样品置检测器中,在15 kV 的加速电压下观测。结果( 图2) 显示,球磨产物的外观形态均呈无规则的块状分布。可见1 与包合载体的混合体系经机械球磨后,体系的微观形态呈无定形分布。

2.2.3 差示量热扫描分析
用差示扫描量热仪测定球磨产物的热力学行为
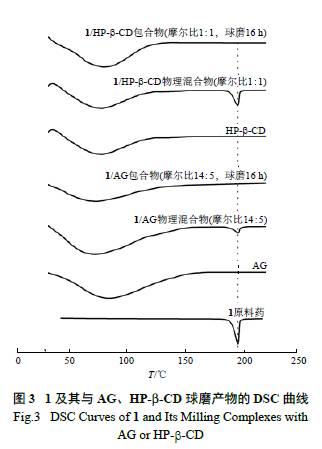
变化。取待测样品5 ~ 7 mg,置具盖的铝盘内,压紧密封;然后,将样品置检测器中,以10 ℃ /min的速率从20 ℃升至250 ℃,Ar 全程作为吹扫气体。1 及其与包合物的热力学行为见图3。1 在195.3 ℃处有一吸热峰,吸热焓为232.5 J/g, 基于Sathigari 等报道[16],判断该吸热峰为1 熔融峰。在1 与AG 或HP-β-CD 的物理混合物中,195.3 ℃处仍然可以观测到1 熔融峰;但1 与AG 或HP-β-CD的包合物中,1 的熔融峰完全消失。基于Mura 等对异丁普生( ibuproxam) 与聚乙二醇4000 复合物热力学行为的分析[17],推测本试验所制备球磨产物的DSC 曲线中,1 熔融峰消失是由于包合物中的药物分子被高度分散并完全被包合在载体材料中,当包合物中的分散介质加热至熔融状态时,药物分子与包合载体一并融化,进而在DSC 的曲线中观测不到原料药的熔融峰。
2.2.4 X- 射线粉末衍射
采用X- 射线粉末衍射仪获取球磨产物的晶体结
构信息。X- 射线源为铜靶Kα 射线(λ=0.154 05 nm),连续扫描的角速度为2 o/min, 对样品的扫描角度为0 ~ 60o( 衍射角2θ)。
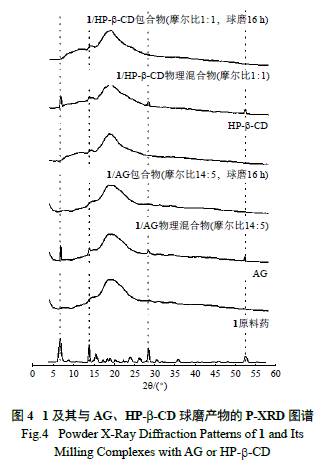
1 原料药及其与包合物的X- 射线衍射行为见
图4。1 原料药的特征衍射峰处于6.5、13.7、15.5、28.2 与52.4o。在1 与AG 或HP-β-CD 的物理混合物中,仍然可以观察到1 处于6.5、13.7、28.2 与52.4o 处的强特征衍射峰,但其处于15.5o 处的弱特征峰被包合载体的宽衍射峰覆盖。在1 与2 种载体的球磨产物中,1 强特征衍射峰已完全消失。该现象可从如下2 个方面进行分析:1) 1 晶状结构在机
械球磨过程中受机械力的作用被破损;2) 1 分子结
构被包裹在AG 或HP-β-CD 的分子骨架中,导致球磨产物中1 特征衍射峰消失。
2.2.5 1H NMR 谱图的检测
通常采用两相体系解释核磁共振技术的弛豫
现象,即假设体系中存在“束缚相”和“自由相”,大部分化合物在其水溶液中呈自由状态,即以“自由相”的形式存在;但是当化合物与周围环境中的其他分子存在分子间作用的时候,化合物分子的运动受到限制,即以“束缚相”的形式存在[ 18]。化合物的弛豫时间分为纵向弛豫时间(T1) 与横向弛豫时间(T2)。在同一体系中,弛豫时间对分子的流动性呈正相关[19—20]。
本课题将1 结构中的甲基质子( δ 3.94) 作为
1/AG 体系与1/HP-β-CD 体系中弛豫时间的测试对象。1 与AG 或HP-β-CD 体系的T2 值分布见图5。1/AG 体系中,T2 值依序为包合物(78 ms)< 物理混合物(212 ms)
定性优于其物理混合物。1/HP-β-CD 体系中,T2 值
依序为包合物(46 ms)< 物理混合物(177 ms)

2.3 1 包合物的溶解度和体外溶出试验
2.3.1 1 含量的HPLC 检测法
2.3.2 含量测定
2.3.3 溶解度的测试
称取不同球磨时间所得的包合物( 约含1
30 mg),各置25 ml 具塞锥形瓶中,加入去离子水10 ml;然后将上述混悬液于37 ℃、200 r/min 振摇1 h;最后经0.45 μm 水膜过滤器过滤后进行HPLC检测,由标准曲线计算药物的溶解度。平行测定5 次。1/AG 体系与1/HP-β-CD 体系中不同球磨时间段的球磨产物及其对应的物理混合物的溶解度见图6。在1/AG 体系中,物理混合物溶解度为0.90 g/L,是1 原料药(0.77 g/L) 的1.2 倍;不同球磨时间产物中,最佳溶解度为1.32 g/L,其球磨时间为8 h。在1 与HP-β-CD 的体系中,物理混合物的溶解度为1.17 g/L;不同球磨时间产物中,最佳溶解度分别为2.16 g/L,其球磨时间为16 h。

2.3.4 相溶解度的测试
分别向不同浓度(0、2、4、8、10 和20 mmol/L,
20 ml) 的HP-β-CD 溶液中加入过量1,然后将上述混悬液于37 ℃、200 r/min 振摇1 h; 最后经0.45 μm 水膜过滤器过滤后进行HPLC 检测,由标准曲线计算药物的溶解度。平行测定5 次。同法在25 和42 ℃条件下平行测定5 次。
根据Higuchi 等报道的方法[ 21],建立了1 与
HP-β-CD 的相溶解度曲线( 图7),确定二者的最佳摩尔比例。相溶解度曲线在25、37 与42 ℃的等温线比较接近,1 溶解度与HP-β-CD 的浓度呈线性增长的关系(AL 型)。根据Brewster 等报道[22],AL 直线的斜率大于1,可能是多个药物分子包合在一个载体材料的分子中;AL 直线的斜率小于1,则可能1 个药物分子包合1 个包合载体分子中。本试验的3 条等温线的AL 斜率均接近0.32,即1 与HP-β-CD 分子形成1 ∶ 1 的包合物。依据方程②~⑤计算得包合物的稳定常数Ks 与其热力学常数( 表1)。包合物的稳定常数显示1 与HP-β-CD 间具有较高的“分子间络合力”,形成的复合物稳定性较高;该体系的热力学参数显示1 与HP-β-CD 的包合可以自发进行( ΔG<0),并且该包合过程是一个熵值增加(ΔS>0) 的过程。



2.3.5 溶出度试验
照中国药典2015 年版四部通则0931 第二法
进行。取包合物适量( 含1 250 mg) 及1 原料药250 mg,分别置溶出杯中;以0.05 mol/L 磷酸二氢钾溶液(pH 6.8)900 ml 为溶出介质,转速为50 r/min,温度为(37±0.5)℃;定时取样品液5 ml( 同时补加同温等量溶出介质),立即用0.45 μm 水膜过滤器过滤,取续滤液进行HPLC 测试,由标准曲线计算药物浓度与药物的溶出率。每个样品进行6 次平行试验。
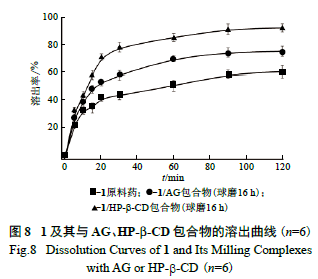
1 及其包合物的溶出曲线见图8。1 的包合物体
系中,第5 min 时,1/AG 体系中药物溶出率相比于原料药略有提高;10 ~ 30 min,1 溶出率提升比较明显,此阶段包合物的药物溶出率明显优于原料药;第90 min 时,包合物体系及原料药的溶出率基本达到平衡;第120 min 时,原料药的溶出率为60%,其1/AG 包合物体系的溶出率为75%。1/HP-β-CD 包合物体系的溶出率类似于1/AG 包合物
体系,第120 min 时,体系的溶出率为92%。本试
验制得1 的包合物体系中,以HP-β-CD 作为包合载体制备的包合物的溶出行为明显优于以AG 为载体的包合物的溶出行为,该结果与溶解度测试结果一致。
2.4 1 包合物体内释药性质考察
2.4.1 试验动物及分组
2.4.2 给药方案及样本采集
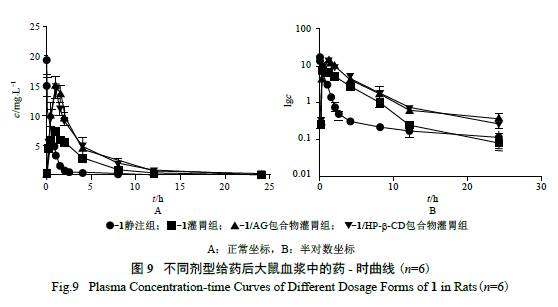
本试验首先按照生物样品分析的相关规范进行
了方法学的确证,结果显示血浆中1 的线性范围在3.6 ~ 26 160 ng/ml,准确度和精确度均<10%,本法符合化学药物临床前药代动力学研究的技术要求。血浆样本检测结果采用DAS 2.0 软件非房室模型计算药动学参数( 见表2),比较不同制剂与原料药在大鼠体内的过程。药- 时曲线见图9。大鼠灌胃给予3 个剂型后,达峰时间tmax 均为1 h,不随剂型的改变而发生明显变化;1/AG 包合物和1/HP-β-CD包合物的cmax 及AUC0 → t 均明显高于1 原料药( 表2)。对比静脉注射1 原料药(10 mg/kg) 与3 种剂型
灌胃给药(50 mg/kg) 的AUC0 → t 值,计算得到1 原
料药、1/AG 包合物和1/HP-β-CD 包合物的绝对生物利用度分别为56.94%、81.63%和78.48%。可见将1 制备成包合物后,其口服生物利用度均得以明显升高。

3 讨论
综合粒度分布、SEM、DSC、P-XRD、NMR
弛豫试验的测试结果,1 与其包合载体经机械球磨处理后,粒径减小;以无定形的形式高度分散在水溶性包合物载体中,并且原料药在该体系中的热力学行为发生一些改变;在水溶液中,药物分子的运动受其包合载体的束缚,证明球磨产物中两组分间存在分子间作用力;即一种不同于物理混合物的新固态形式在机械球磨过程中得以形成。1 经球磨后的产物与包合物体系的行为相吻合,即球磨产物的新固态形式为包合物。
在体外溶出试验中,1/HP-β-CD 包合物与1/
AG 包合物均有效地提高了1 溶解度及其溶出速率,是因为包合物体系中的分子间作用力促进了难溶性的药物分子较好地溶解在水中。在1/AG 包合物的溶解度试验中,球磨时间8 h 的包合物优于球磨时间16 h 的包合物,该现象说明这种分子间的作用力具有饱和性,并且长时间的球磨会减弱该分子间作用力。体内试验显示:口服1/AG 包合物的绝对生物利用度为原料药的1.43 倍,口服1/HP-β-CD 包合物为原料药的1.38 倍。以AG 为载体的包合物生物利用度略高于以HP-β-CD 为载体的包合物,该现象与AG 具有一定的血管通透特性有关,在体内AG 包载着1 通过胃肠道黏膜进入血液,促进了1在胃肠道的吸收,进而提高1 的生物利用度。
本试验采用滚动式球磨仪成功地制备了1/AG
包合物和1/HP-β-CD 包合物。体内外试验表明:2种包合物体系中,载体并不影响其原料药的理化特征,但却能改善1 溶解度及溶出行为,提高其口服吸收生物利用度,为后续开发低药物剂量、高口服吸收生物利用度的1 剂型提供了试验基础。
作者简介:张理星(1967—),男,研究员,主要从事药物研发及工
程化研究。
Tel:0539-5030990
E-mail:[email protected]