
自然界中的功能结构通常由特定的几何单元结构精确组装而成,设计和构建目标超分子结构很大程度上也取决于如何合理安排单元结构中官能团的空间位置。利用金属离子结合多齿鳌合的有机配体进行超分子组装受到越来越多科学家的重视,基于基本结构单元自组装、自互补、自排序等技术,通过调控不同配体的立体电子效应,人们可以设计出一系列高度对称的功能结构。
获得2016年诺贝尔化学奖的“分子机器”是指由分子尺度的物质构成、在外界的刺激下能行使某种特殊功能的器件,“分子镊子(molecular tweezer)”是其中有趣的一种结构。作为一种分子受体,分子镊子含有两个呈顺式构象连接的相同“平臂”。“平臂”通常由含有芳香烃的分子组成,并通过中间的刚性连接体控制两“平臂”的相对空间取向。通常情况下,两“平臂”相互作用位点的距离大约为7 Å,通过不同芳香烃底物与镊子“平臂”的π堆积作用实现两者的结合。在分子镊子中引入金属离子则可以为发展此类超分子结构提供更多的思路,相比于有机分子间的相互作用,金属离子结合有机配体可以形成取向更为明确的结构,从而大大简化合成的方法。
近日,西班牙海梅一世大学的Eduardo Peris教授课题组设计了一种引入Au(I)金属离子的分子镊子,这种结构可在多种不同金属阳离子的驱动下,结合π-π堆积作用发生自组装,形成具有特殊空腔的二聚体,由此实现分子镊子的功能。相关工作发表在Angew. Chem. Int. Ed. 上。

Eduardo Peris教授。图片来源:Universitat Jaume I
Au(I)络合物可以和芳香炔类化合物形成稳定的线型分子,广泛应用于寡聚物、高分子材料的合成中。此外,炔基-金片段还可通过亲金相互作用自组装形成线型的超分子结构。作者设想将这种线型结构运用到设计分子镊子的“平臂”中,合成路线如下图1所示,1,8-二乙炔基蒽和芘稠合的氮杂环卡宾金络合物NHC-Au(I) 1在甲醇的碱性溶液中能以75%的收率得到分子镊子2。核磁共振波谱表征显示,2在CDCl3中以单体的形式存在,而在C6D6中,2会通过π-π堆积作用自组装形成二聚物(2)2。作者还从双炔A与Au(I)络合物2出发,反应中同时加入AgBF4作为氯离子束缚剂,也可以直接形成二聚物3,其中阳离子Ag+与Au(I)金属中心的相互作用促进这一过程的发生。
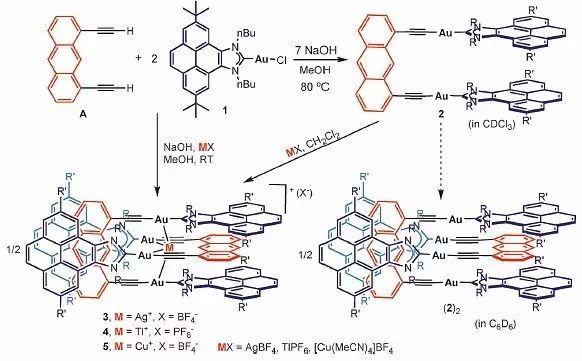
图1. 分子镊子的合成路线以及自组装过程。图片来源:Angew. Chem. Int. Ed.
除此之外,他们发现Tl+、Cu+离子都可以驱动自组装过程形成二聚物4、5,X射线单晶衍射表征分别证实了(2)2、3、4的晶体结构,在(2)2中,四个Au原子构成长方形取向,Au-Au的距离分别是3.32 Å、4.9 Å,蒽和芘的平均距离为3.68 Å。3中Ag和Au的距离为2.86-2.92 Å,4中Tl和Au的距离为3.10-3.15 Å。这些数据证实了Au(I)与不同金属阳离子间存在强相互作用。
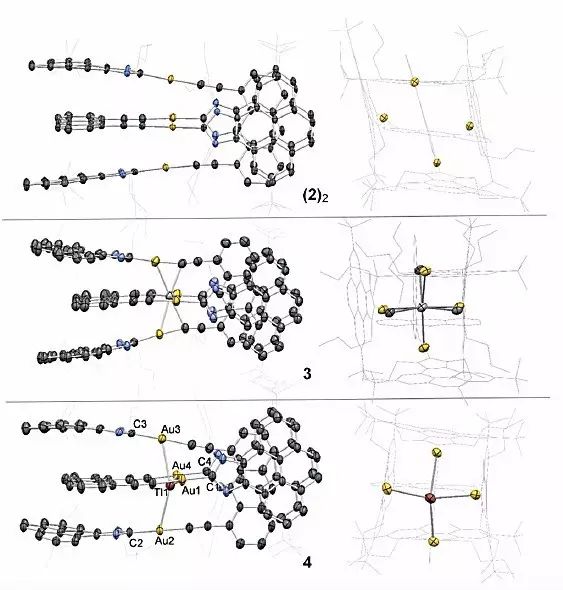
图2. 分子镊子二聚物(2)2、3、4的X射线单晶衍射结构。图片来源:Angew. Chem. Int. Ed.
作者还分别以二氯甲烷作为溶剂,通过2与AgBF4、TlBF6混合定量得到二聚物3和4,从而证实这两种二聚体需要在2的生成反应进行完全后进而发生自组装来实现,并非同时发生。他们还通过核磁共振波谱手段对二聚物的形成过程进行监测,同样证实了以上分步反应的存在。
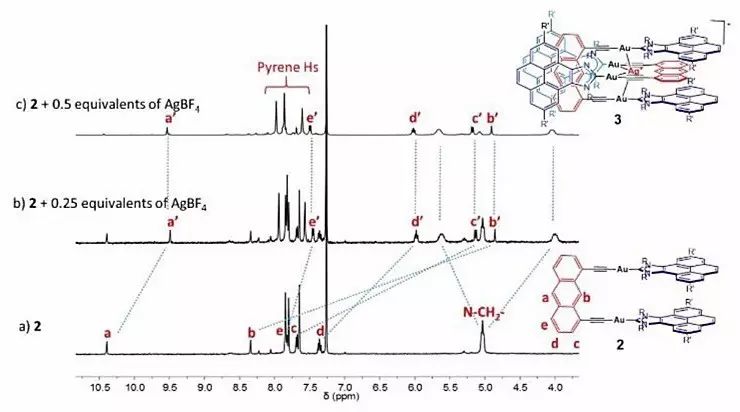
图3. 二聚物3形成过程的核磁共振波谱表征。图片来源:Angew. Chem. Int. Ed.
随后,作者借助紫外-可见和荧光光谱进行滴定实验,进一步证实了二聚物的形成。与此同时他们也测出Ag+、Tl+、Cu+三种阳离子驱动分子镊子2发生自组装的结合常数分别为2.7 x 109, 4.2 x 108, 7.9 x105 M-2,这一结果与ESI-TOF-MS质谱竞争实验的测试结果相吻合(Ag+ > Tl+ > Cu+)。
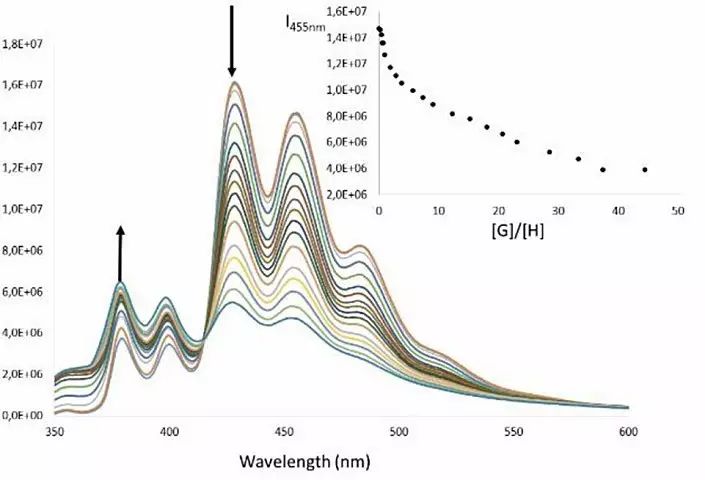
图4. 2与Tl+混合形成二聚物4的荧光光谱滴定实验。图片来源:Angew. Chem. Int. Ed.
——总结——
Eduardo Peris教授报道了一种含有Au(I)金属离子的分子镊子,这种结构可在多种不同金属阳离子的驱动下发生自组装,形成具有特殊空腔的二聚体,二聚体的结构通过X射线单晶衍射、核磁共振波谱以及质谱等表征手段进行了佐证。 π-π堆积和金属间相互作用在自组装过程中至关重要。
原文(扫描或长按二维码,识别后直达原文页面):

Cation-Driven Self-Assembly of a Gold(I)-Based Metallo-Tweezer
Angew. Chem. Int. Ed., 2017, 56, 9786, DOI: 10.1002/anie.201704359
(本文由PhillyEM供稿)


本文版权属于X-MOL(x-mol.com),未经许可谢绝转载!欢迎读者朋友们分享到朋友圈or微博!
长按下图识别图中二维码,轻松关注我们!
点击“阅读原文”,查看所有收录期刊











