莱伯氏先天性黑朦是一种常染色体隐性遗传病,是导致儿童失明的主要遗传疾病,目前无任何上市疗法。随着基因治疗相关技术的发展,莱伯氏先天性黑朦患者终于有了被治愈的希望。生物医药公司们正为此努力着。
症状——视力恶化至失明
患者视力通常从婴儿时期开始持续恶化,并最终失明。主要病理特征是患者眼睛后方用于感知光线和色彩的视网膜细胞的逐渐退化,症状包括眼球震颤、畏光、瞳孔散大且反应迟钝、圆锥性角膜、极度的远视等。另外,患者常有按压或揉戳眼睛的行为。
患者分布——他们是散落在人群中的小群体
患者分布无明显的地域性偏好。每10万新生儿中,就有2-3人得病。按此发病率计算,中国有三万余名患者。
发病原因——各不相同
以下20余种基因任意一个发生突变,都有可能引起莱伯氏先天性黑朦。其中CEP290, CRB1, GUCY2D, RPE65这四种基因的突变最为常见。
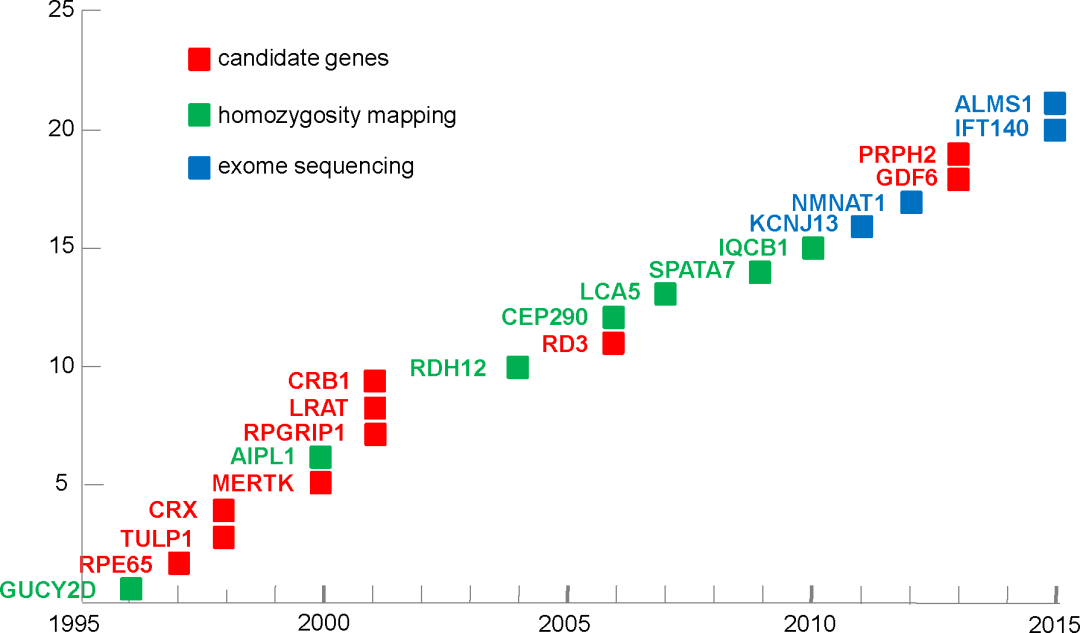
(通过不同方法不断鉴定出的莱伯氏先天性黑朦相关基因)
这已知的20余种基因突变仅占莱伯氏先天下性黑朦患者的约70%。也就是有约30%的该病患者的病因还没有被找到。因此,这近30%的患者的治疗和预防都毫无头绪。
目前针对最常见的两种基因型的莱伯氏先天性黑朦RPE65(约占所有该病患者的16%)及CEP290(约占所有该患者的30%),均已有疗法进入商业化开发阶段,正式上市指日可待。其它多种基因型也有治疗方法进入临床前期研究阶段。
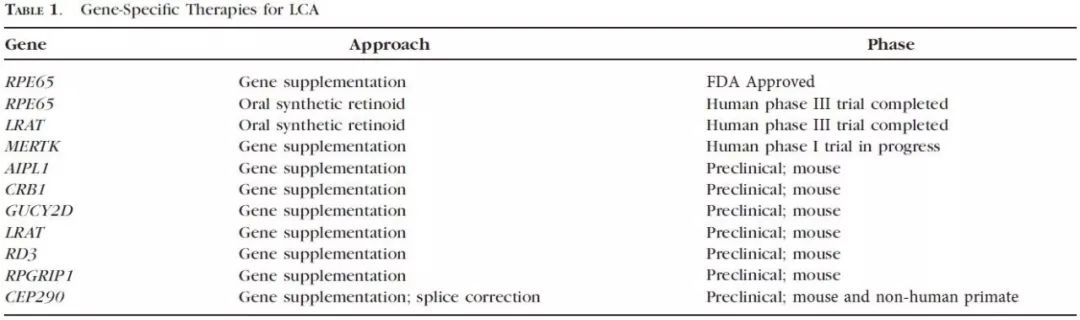
(目前经过概念验证的基因疗法研发进程及相对应的莱伯氏先天性黑矇基因型)
以下是正在进行商业化的几个代表性疗法:
针对RPE65型莱伯氏先天性黑朦的根治性疗法通过FDA评审
疗法:
LUXTURNA
发明者:
SPARK Therapeutics

适应症:
1岁以上,双等位基因RPE65突变,且眼底保有足够视网膜色素上皮细胞的莱伯氏黑朦患者。
研发阶段:
通过FDA评审,等待上市。
疗法类型:
基于AAV(腺相关病毒)的基因添加疗法。在视网膜色素细胞等非分裂细胞中理论上可终生发挥作用。
2017年10月13日,首款能够矫正人类基因缺陷的疗法——Luxturna正式通过FDA评审,拟于2018年上市,预期定价100万美元。该款疗法针对的正是莱伯氏先天性黑朦的RPE65基因。该基因病变患者占所有莱伯氏黑朦患者的16%。他们眼底的视网膜色素细胞因无法正常表达PRE65基因而退化,从而导致失明。
Luxturna的本质是携带健康RPE65基因的AAV(腺相关病毒)。向视网膜下注射Luxturna,AAV会吸附在视网膜色素上皮细胞上,并将携带有RPE65基因的腺病毒基因组注入这些细胞,从而持续表达正常的RPE65基因,替代突变的RPE65发挥功能,延缓或抑制视网膜色素细胞的退化。
Luxturna疗效较为显著持久,无明显副作用。一期临床实验至今已4年,12名儿童视力均得到了提升,且无明显相关副作用。三期临床持续一年多以来,经治疗的65%(13/20)患者均顺利通过了最低照度测试,即在极暗的光线下顺利通过了一条充满障碍的道路。目前无复发,且同样无明显相关副作用。
RPE65及LRAT型莱伯氏先天性黑朦口服药结束二期临床
疗法:
Zuretinol
发明者:
Novelion Therapeutics

适应症:
RPE65或LRAT突变型莱伯氏先天性黑朦及色素性视网膜炎
研发阶段:
2期临床,临床1b期结果显示,70%患者视力得到提升
疗法类型:
口服维生素补充剂,须长期服用
正常的视色素细胞受到光刺激后将11-顺式视黄脂(一种维生素A)转变为全反式视黄脂,产生电信号,传导至视神经元,后全反式视黄脂恢复为11-顺式视黄脂再次参与感光及信号传递。RPE65及LRAT编码的蛋白在这个循环过程中起着非常重要的作用。RPE65及LRAT型先天性黑朦患者正是由于基因突变使得这两种基因的蛋白表达异常,使得该循环过程无法持续,从而造成视力损伤。
Zuretinol本质是合成9-順式-维生素A醋酸酯,它可以替代缺失的的视网膜11-顺式视黄脂,起到提高视力的作用。
CE
P290型莱伯氏先天性黑矇疗法EDIT-101临床试验即将启动
疗法:
EDIT-101
发明者:
Editas Medicine及Allergan Pharmaceuticals International联合开发及商业化

适应症:
CEP290 (c.2991+1655A> G) 内含子双等位基因突变的患者。
研发阶段:
临床前研究,预计2018年中旬进入1/2期临床试验。已成功完成非人类灵长类动物及人源化小鼠实验,基因编辑效率可达50%。正在进行为期一年的CEP290患者病程观察,为临床试验确定适应症患者。
疗法类型:
基于CRISPR的基因编辑治疗
CEP290基因突变是莱伯氏先天性黑朦最常见也是最严重的类型,占所有该病征患者的15%-22%,也有报告指出为30%。尤其是其中的(c.2991+1655A > G)突变就占到了所有莱伯氏先天性性黑朦患者的15%。该基因突变主要是因为内含子中的一个突变导致CEP290转录出的mRNA中含有一个提前终止密码子,导致CEP290翻译的蛋白功能不全。而这种蛋白又直接影响了视网膜光感细胞的发育和功能,导致严重的视力损伤。
针对这一突变,Editas开发出名为EDIT-101的疗法。其本质是装载有CRISPR-Cas9蛋白及两个sgRNA的AAV病毒。将EDIT-101注射入视网膜下,AAV会将其基因组注射入光感细胞,从而在这些细胞中表达CRISPR-Cas9酶和sgRNA。这两个sgRNA会将Cas9酶准确地引导至突变位点的两侧。Cas9在此两个位点制造双链断裂,将突变位点拿掉,从而使CEP290转录出的mRNA恢复正常,最终恢复患者视力。







