
撰文 | 紫薯
2017年6月9日
● ● ●

肿瘤融合基因突变靶向药物最新进展
引言
最近,
Loxo Oncology
因为最新公布的
TRK
融合蛋白小分子激酶抑制剂的积极
”
篮子
“
临床试验数据,成为
ASCO
和国内外媒体热捧的明星。那么肿瘤融合突变是怎么发现的,还有其它的什么类型,究竟占多少比例,最新的药物研发进展如何?我们在这里小结一下。
融合基因是从两个先前分开的基因而形成的杂合基因。融合基因的产生主要有三种情况:
易位
、
中间缺失
,或
染色体倒位
。
最早的有关融合基因的报道来是由
Peter Nowell
和
David Hungerford
于
1960
年检测到的慢性粒细胞白血病
(CML)
的染色体异常,后来被指定为费城染色体
[1]
。
1973
年
, Janet Rowley
发现费城染色体突变实际上是由
9
号和
22
号染色体上的两个片段融合形成的,而不是之前认为的
22
号染色体缺失突变造成的
[2]
。到了
80
年代初,人们才发现融合突变形成了新的杂合基因
BCR-ABL
。到
1985
年,证实了
BCR-ABL
是
CML
的主要致癌基因突变
[3]
。接下来,就是诺华开发的迄今为止最成功的靶向激酶小分子抑制剂格列卫(
Gleevec
),把致死率曾经极高的
CML
变成了慢性病
[4]
。
最早在实体瘤中发现的融合基因之一是
TPM3-NTRK1 (
编码
TPM3-TRKA
融合蛋白
)
,于
1986
年从结肠癌组织样品中克隆得到并报道
[5]
。直到
2014
年,
TPM3-NTRK1
融合突变才再次在结肠癌样品中被重复发现
[6]
。
Fusion genes and their discovery methods
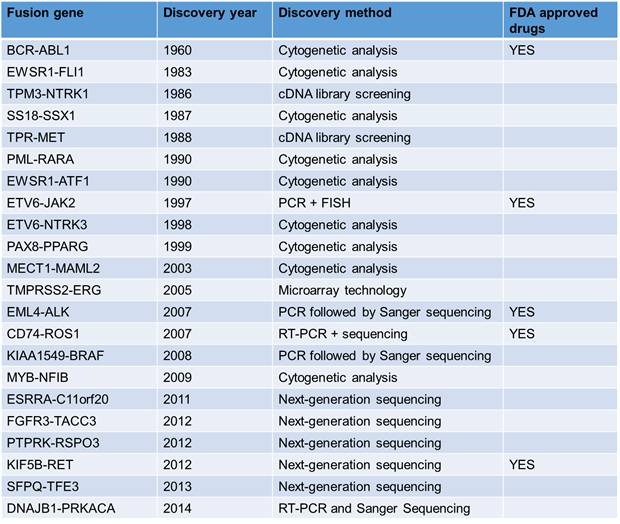
在
2000
年前,总共大约有
600
个融合基因被发现,主要集中在血癌样品中。肿瘤融合基因发现的数量随着基因测序技术的发展完善在
2014
年达到高峰,当年报道了超过
7854
例融合基因,而在此之前,总共发现的融合基因还不到
2000
例,而最近两年新发现的融合基因数量也开始大幅减少,显示多数融合基因可能已经被发现
[7]
。迄今为止
(
截止日期
5-12-2017)
,人们总共发现了超过
10,861
个癌症相关的融合基因突变
(Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer) [8]
。融合突变分布于几乎所有的癌症类型。肿瘤融合基因突变不仅在诊断和预后过程中起重要的指导作用,还逐渐成为一个有效的靶向药物研发对象
[9]
。
根据功能分类,肿瘤融合基因可以分为以下几类:
激酶类、转录因子类、代谢酶类、
Wnt
信号通路类、
TGFβ
类、染色质修饰基因等
[10]
。所有这些基因都具有原癌基因属性。它们功能的过度激活均是重要的致癌因素。而融合基因通过二聚体化等修饰,使得这些原癌基因变得持续激活,不受控制,从而成为致癌主导因素。
Discovery of fusions coincides with improved DNA sequencing technologies
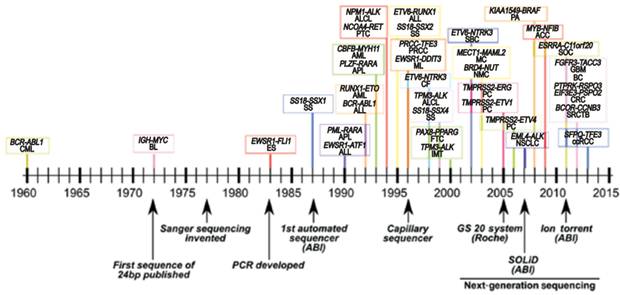
我们这里只重点小结现今可靶向的一些融合突变,也就是激酶类的融合突变。主要结合最近
MSKCC
发布的大规模的晚期肿瘤临床检测治疗数据,来讨论可靶向融合突变的患者比例,以及相关靶向药物研发最新的进展。
继在《癌症发现》上报道了
860
例晚期肺腺癌患者的最新诊断,治疗数据之后
[11]
,美国最好的、最大的癌症中心之一,纪念斯隆凯特琳癌症研究中心
(MSKCC, MSK)
进一步在《自然医学》杂志上公布了其对超过万名晚期癌症患者的基因测序及个性化诊断数据,开启了崭新的癌症检测治疗模式
[12]
。
MSK
使用的二代测序技术,
MSK-IMPACT
(
Integrated Mutation Profiling of Actionable Cancer Targets
)是一个更加全面的组合癌变基因测序方案。起初是基于
341
个癌变基因的组合,最近增加到
468
个。文章报道的患者数量为
10336
名,是截止到去年夏天的数据。最新的患者数据已经增加到超过
16000
名。而且,数据库还在以每月
600-800
名患者的数量在增长。(数据库地址
http://cbioportal.org/msk-impact
)
与传统的全外显子测序相比,
MSK-IMPACT
对基因上所有的重要区域都进行了测序,可以检测到基因上的蛋白编码区突变、拷贝数变化、启动子突变和基因组重排。
MSK-IMPACT
能够检测到更详尽的,更全面的基因融合突变。研究人员在
1597
名患者中都检测到了融合基因突变,
占所有患者的
15%
。其中最常见的分别是
TMPRSS2-ERG
、
EGFRvIII
、
EML4-ALK
,
EWSR1-FLI1
等。
在所有融合突变中,有
35%
是与激酶相关的突变
(n=268
),约占三分之一。由于激酶融合突变大多数是功能持续获得性突变,使得激酶融合突变成为一个有效的致癌突变指标和靶点。
有些激酶融合突变多集中在特定的肿瘤类型,如
EML4-ALK
、
CD74-ROS1
,主要集中在非小细胞肺癌(
NSCLC
),
KIF5B-RET
相关的突变主要集中在
NSCLC
和甲状腺癌,
FGFR2
的融合突变主要发生在胆管癌,
FGFR3
融合突变主要集中在神经胶质瘤。而另外一些融合突变则比较均匀的分布在不同的肿瘤类型,如
BRAF
、
NTRK1
、
NTRK3
等。同时,
MSK-IMPACT
数据还揭示了
51
个新的融合组成片段。
大多数情况下,激酶融合突变是主要的单一驱动致癌突变,与其它驱动突变相互排斥。而普通的基因点突变很多情况下是非驱动突变。如文章中报道的
ROS1
融合突变有
29
例,(最新更新的
ROS1
融合突变有
42
例),而相应的
ROS1
点突变等有多达
432
例。绝大多数
ROS1
的点突变不是驱动致癌突变,也不能用来作为靶向药用药标准。类似的情况发生在
ALK
、
NTRK1
,
2
,
3
、
RET
等基因突变上。(当然,也有许多点突变是驱动突变的例子,如
EGFRL858R
突变是非小细胞肺癌的主要驱动突变之一,
BRAF V600E
驱动突变占黑色素瘤的一半。)
Spectrum of kinase fusions identified by MSK-IMPACT
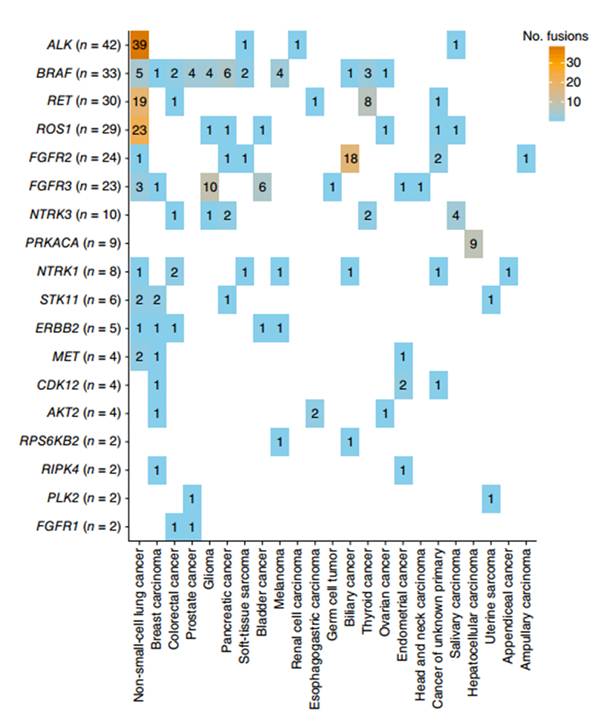
这里,我们总结一下肿瘤可靶向激酶融合突变的药物研发进展,以及大规模测序对个性化药物研发和临床试验的积极推动作用。主要介绍万人测序中超过
10
个患者的激酶融合突变,这使得临床试验变得更加可行。按人数多少排序,包括
ALK
、
BRAF
、
ROS1
、
RET
、
FGFR1-4
、
NTRK1-3
。有些靶点前文已经更新过,这次重点小结以前没有提到的靶点,如
FGFR1-4
和
NTRK1-3
。
EML4-ALK
融合突变
ALK
的融合突变最早报道于
1999
年,变性大细胞淋巴瘤中的
TPM3-ALK [13]
。但
1997
年发表在《
Nature》
上的有关非小细胞肺癌
EML4-ALK
的融合突变的报道才真正引起了人们的重视,开启了固体瘤融合突变靶向药研发的热潮
[14]
。
ALK
的驱动突变主要是
EML4-ALK
融合突变,集中发生在较年轻
(
平均年龄
52
岁,肺癌总体发病平均年龄为
70
岁)的非吸烟人群中,约占非小细胞肺癌的
4-7%
。
MSK-IMPACT
在总共发现的
42
例
ALK
融合突变患者中,有
39
例发生在非小细胞肺癌,在万人晚期肿瘤检测中的比例为
42/10336=0.4%
。这个比例在所有可靶向激酶融合突变中已经是最高的了。美国每年
ALK
融合突变的患者大约在
9000-10000
人。我们可以用这个作为基准,来比较其它融合突变的多少。
辉瑞的
Crizotinib
是最早获得
FDA
批准的
ALK
小分子抑制剂,同时也是首次获批的固体瘤融合突变靶向小分子酪氨酸激酶抑制剂
(TKI)
。后来诺华的
Ceritinib
,罗氏的
Alectinib
,以及最近获得加速审批的
Ariad
的
Brigatinib
均是在
Crizotinib
之后的二代,二线靶向药,可以有效克服
Crizotinib
的耐药性。最新刚刚获得突破性疗法的辉瑞的
ALK TKI
三代
Lorlatinib
的
ALK
激酶活性最高,能够较有效地克服一代和二代
ALKTKI
的耐药性。在临床上表现活跃还有贝达的
Ensartinib
,和
TP Therapeutics
的
TPX-0005 [15]
。
ALK
inhibitors in clinical trials
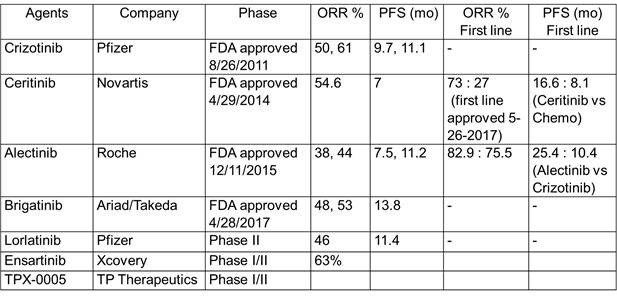
Crizotinib
的
ALK
活性相对较低,而相应的应答持续时间也较短,二线
ALKTKI
的活性得到大幅提高,争当一线疗法的临床试验是最新的
ALK TKI
热点之一。诺华的
Ceritinib
已经完成三期临床,与化疗相比较,数据积极,获得
FDA
批准用于
ALK
初始患者一线用药
(5-26-2017)
。罗氏的
Alectinib
在日本的三期临床试验,头对头与
Crizotinib
比较,效果好一倍还多。
Ariad
的
Brigatinib
及辉瑞的
Lorlatinib
均在进行三期临床试验,争取将来推到一线使用。
最新的
Alectinib
的美国
(
全球
)
临床试验数据显示
(2017
,
ASCO # LBA9008) [16]
:与
Crizotinib
头对头比较,
Alectinib
能够降低死亡危险超过
53%
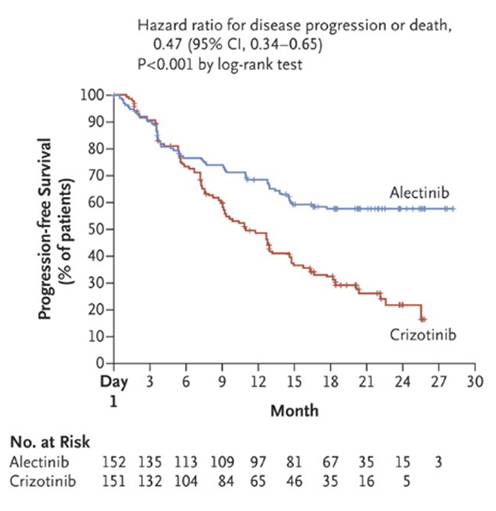
。中位无进展期(
PFS
)
25.4 vscrizotinib 10.4
个月,提高了一倍多。而且中位
PFS
还没有完全达到,证实了之前在日本进行的临床试验。
这是一个巨大的提高,也是迄今为止见到的实体瘤小分子激酶抑制剂的最好成绩。同时说明,靶向激酶小分子的潜力还很大,可提升的空间还可以非常可观
。在
Ceritinib
的
PFS Kaplan-Meier
生存曲线上,我们也看到了只有在免疫疗法中看到的
”
平尾
“
现象,表明相当比例的患者病情得到长期的控制。同时,这个平尾发生在应答率
50%
以上,尤为可贵。
Alectinib vs Crizotinib PFS
BRAF
融合突变
BRAF
的驱动致癌突变主要是
BRAF V600E
点突变,在黑色素瘤中占到
50%
的比例。
BRAF
的融合突变比例相对较少,但从
MSK-IMPACT
的数据看,
BRAF
在最多的肿瘤类型中检测到
(n=33)
。这个比例(
0.3%
)与以前报道的数值相当
FoundationOne Heme™ comprehensive genomicprofiling assays (n=55/20573
,
0.3%
)
[17]
。
虽然靶向
BRAF V600E
的
TKI
已经在黑色素瘤上获得
FDA
批准,但在
BRAF
融合突变中还没有系统的临床研究。
RET
融合突变
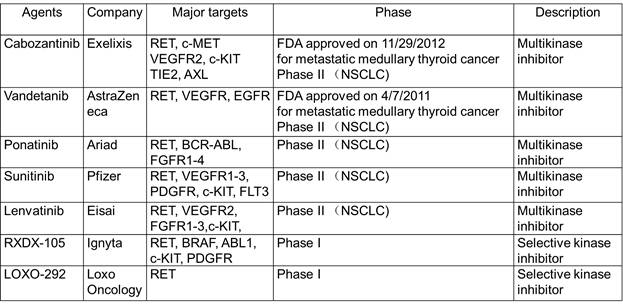
RET
的融合突变总数比
ROS1
还多一例!达到
30
例。其中多数集中在
NSCLC
,有
19
例。而甲状腺癌中也有较高的比例
(8
例
)
。前文介绍过
MSKCC
的
Dr. Drilon
主导进行的
Cabozantinib
在
RET
融合突变中的临床试验
[18]
,总体应答率
27%
,与其它
TKI
比相对偏低。个人认为主要原因可能是
Cabozantinib
的活性不够高,而
VEGFR
的靶向毒性限制了其剂量使用。其它的多靶点的激酶小分子抑制剂活性比
Cabozantinib
还低,预期效果不会更好。
Cabozantinib
在
2012
即获得
FDA
批准,用于治疗转移性甲状腺髓样癌,一小类罕见的含有大量
RET
突变的甲状腺癌。但换到非小细胞肺癌上,
Cabozantinib
的疗效窗口就显得非常吃力。也说明,组织差异性有时是真实存在的。
Ignyta
和
Loxo Oncology
均设计了选择性较高的
RETTKI
,活性也有所提高,期待他们的临床表现。
RET inhibitors in clinical trials
ROS1
融合突变
ROS1
融合突变在
NSCLC
中大约占
1.5%
。
MSK-IMPACT
检测到
29
例
ROS1
融合突变(最新数据有
42
例),其中
23
例发生在
NSCLC
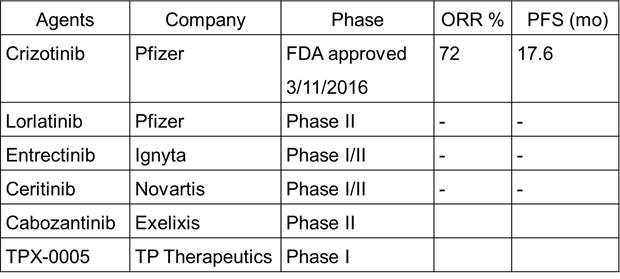
,6例发生在其它肿瘤类型。凭借50例ROS1融合突变患者的积极临床数据获得FDA加速批准的Crizotinib有较好的ROS1 活性,患者中位PFS达到17.6个月,是Crizotinib在ALK融合突变患者上的两倍[18]。这个显著较长的应答究竟是由ROS1靶点本身决定的,还是因为Crizotinib有更好的ROS1抑制活性,尚无定论。
Ignyta的Entrectinib和诺华的Ceritinib也有不错的ROS1抑制活性,但他们不能有效地克服Crizotinib一线治疗产生的耐药突变,如solvent front突变等。所以它们也主要在进行初始(Naïve)患者的临床试验。
Exlelixis的Cabozantinib,辉瑞的Lorlatinib,TP Therapeutics 的TPX-0005具有更高的ROS1活性,能够克服Crizotinib的耐药突变,相关的临床试验正在进行中。
ROS1 inhibitors in clinical trials
FGFR
融合突变
FGFR
家族包括
4
个
FGFR
受体亚型,
FGFR1-4
,多达
18
个
FGF
生长因子配体。与上面提到的
ALK
,
ROS1
,
RET
等相比,
FGFR1-4
在成人体内起着更重要的生理功能,包括组织、代谢平衡、内分泌、血管生成、伤口愈合等。也意味着靶向
FGFR
可能有更多的靶点直接相关的毒性
[19, 20]
。
FGFR1
的融合突变比较少见,
MSKCC
文章中只有两例报道,更新的数据有
5
例。
FGFR1
更主要的致癌因素是过量表达。
FGFR2
的融合突变主要集中在胆管癌。文中共报道了
24
例
FGFR2
融合突变,
(
最新数据
35
例
)
,在总共
242
例胆管癌中有
18
例
FGFR2
融合突变
(
最新数据
25
例
)
,比例高达
7.4%
。对于
FGFR
小分子
TKI
来说,这应该是一个很好的突破点。
FGFR3
融合突变共报道了
23
例。主要发生在神经胶质瘤,共有
10
例。其次是膀胱癌,有
6
例。
FGFR4
的融合突变在文章中没有报道,但更新的数据显示了两例。
万人数据中,
FGFR1-3
融合突变加起来,总数是
49
例,超过了
ALK
融合突变的比例
!
显然是一个更大的“篮子”。
针对
FGFR
靶点的大分子药物也有报道,如
FGFR2
、
FGFR3
的单抗,
FGF2
的
traps
等,但临床数据还不多。而且,大分子药物对多位于胞内的融合突变也无法起作用。这里主要小结一下
FGFR
小分子
TKI
。
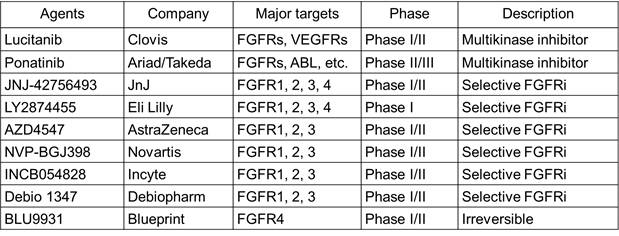
首先是多靶点激酶抑制剂类。因为
FGFR
的激酶结构域与
VEGFR
、
PDGFR
等相似性很高,许多之前针对
VEGFR
、
PDGFR
的小分子
TKI
也多具有
FGFR
活性。如
Ariad
的
Ponatinib
、
Clovis
的
Lucitanib
等。总体来讲,多靶点的
FGFR TKI
的临床疗效不显著,由
VEGFR
引起的靶向毒性作用也限制了
FGFR
的药物用量。我们甚至看不到由靶向
FGFR
引起的靶点直接相关的毒性,如组织钙化,高磷血症等。不看好这类抑制剂在
FGFR
融合突变上的应用。
接下来是选择性的
FGFR
小分子
TKI
。强生的
JNJ-42756493
和
Eli Lilly
的
LY2874455
属于广谱型的
FGFR
小分子抑制剂,能够同时抑制
FGFR1-4
。而更多的是选择性抑制
FGFR1-3
的小分子
TKI
,如诺华的
NVP-BGJ398、
阿斯利康的
AZD4547
等。
Blueprint
还有选择性高的
FGFR4
不可逆的小分子抑制剂,
BLU9931
。
FGFR1-4 inhibitors in clinical trials
由于
FGFR
的致癌因素较多,包括过量表达、点突变、融合突变等,但患者分布却不集中。针对
FGFR
的临床试验病人招募成为一大难题。比如,
FGFR1
过量表达在多种肿瘤中发生,在










