燃料电池是新一代能源转换器件,然而其广泛应用受限于阴极的氧还原反应(ORR)。在商业化质子交换膜的推动下,酸性条件下的氧还原反应是燃料电池发展的突破方向。
现阶段广泛使用的是铂催化剂,其稀有的储量和高昂的价格阻碍了燃料电池的广泛应用。过渡金属基催化剂的应用在很大程度上受限于它们在酸性介质中的低ORR活性和结构稳定性, 因此发展高活性、高稳定性的过渡金属催化剂仍然面临着巨大的挑战。
1、在与氮掺杂碳材料复合的Fe簇中,实现了Fe原子数的精确调控;
2、氮掺杂碳上的Fe2簇在酸性介质中表现出优异的氧还原性能;
3、通过调节Fe簇中的Fe原子数,可以调节氧吸附行为可以由特定数量的调控;
4、从Fe团簇入手,可以确定氮掺杂碳的氮种类和氮含量。
近日,中国科学技术大学熊宇杰教授与杭州师范大学高鹏教授、复旦大学陈萌教授(共同通讯作者)等合作在国际著名杂志Chem发表题为“Precisely Tuning the Number of Fe Atoms in Clusters on N-Doped Carbon toward Acidic Oxygen Reduction Reaction”的文章。
他们通过精准调控Fe团簇所含的铁原子数目,实现了含有不同铁原子数的负载型铁团簇催化剂,在酸性氧还原反应中展现出优异的催化活性。
图1. Fe-N-C催化剂的合成和表征:(A)两步合成Fe2-N-C;(B-E) Fe2-N-C的(B)SEM,(C)TEM和(D)HAADF-STEM图像;(E)EDS元素分布图。
Fe2-N-C的两步合成:在ZIF-8的空腔中原位封装Fe2(CO)9,形成Fe2(CO)9@ZIF-8核壳结构前驱体,经高温热解获得锚定在氮掺杂碳上的Fe2簇(图1A)。
由SEM(图1B)可以看出Fe2-N-C颗粒为菱形十二面体,大小约为200nm。TEM显示在多面体中没有明显的Fe纳米颗粒(图1C)。
HAADF-STEM(图1D)显示出随机分布在N掺杂的碳表面的Fe2二聚体(如图1D中的红色圆圈所示)。当Fe2团簇与TEM中电子束的入射方向对齐时,它们可能在图像上表现为单个亮点。EDS元素分布图表明Fe、C、N元素在样品中均匀分布(图1E)。
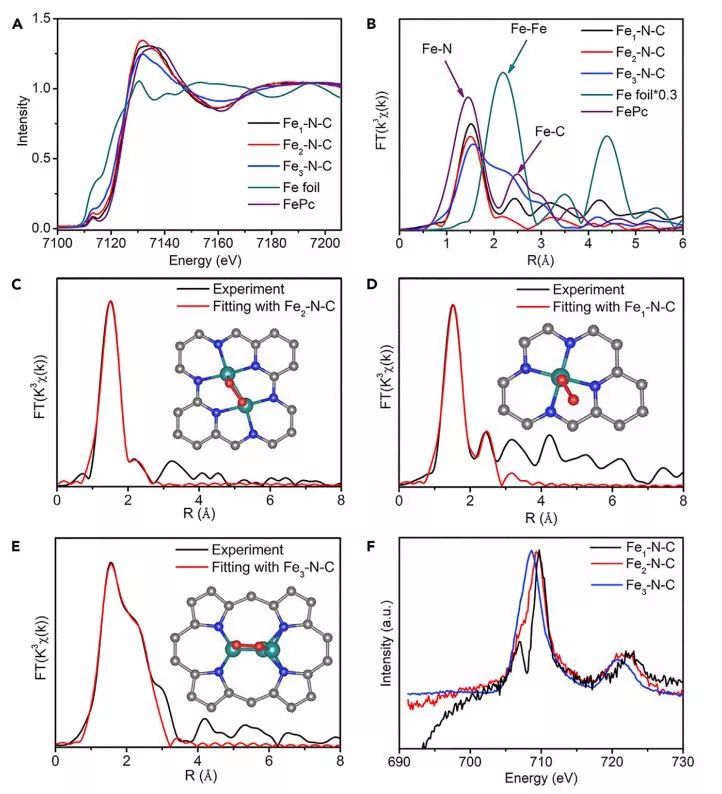
图2. 基于XAFS的精细结构表征:(A)Fe1-N-C,Fe2-N-C和Fe3-N-C的归一化Fe K-edge XANES光谱;(B)k3-weighted傅里叶变换Fe K-edge EXAFS光谱;( C ) Fe1-N-C,( D ) Fe2-N-C和 ( E ) Fe3-N-C的k3-weighted傅里叶变换实验的Fe K-edge EXAFS光谱(黑线)和拟合曲线(红线);(C-E)的插图分别显示了由理论计算得到的Fe1-N-C,Fe2-N-C和Fe3-N-C的优化结构模型(Fe:绿色,N:蓝色,C:灰色和 O红色);(F)Fe1-N-C,Fe2-N-C和Fe3-N-C的Fe L2,3- edge XANES光谱。
图2A显示了Fe1-N-C,Fe2-N-C和Fe3-N-C的归一化Fe K-edge XANES光谱,与Fe箔和FePc相比,Fe1-N-C中不存在位于2.46 A 的Fe-Fe键,表明分离的Fe原子的性质(图2B)。
从EXAFS拟合曲线中提取键长和配位数(CN)来解析成键信息,Fe2-N-C和Fe3-N-C样品的Fe-Fe键的CN分别为1.2和2.4,表明Fe物种主要以Fe2和Fe3簇的形式存在于Fe2-N-C和Fe3-N-C,而不是以Fe基纳米颗粒的形式存在。
随后,该工作通过理论计算模拟了将Fex团簇结合到氮掺杂碳层的优化结构,通过在Fe1-N-C中形成四个Fe-N键,优先将单个Fe原子锚定在碳层上(图2D);而在Fe2-N-C中每个Fe原子与三个N原子和一个Fe键合(图2C);Fe3-N-C中的三角形Fe3簇与碳层不平行,其中每个Fe原子与两个N原子和两个Fe原子键合(图2E)。
Fe L2,3-edge XANES 光谱表明,Fe1团簇需要比Fe2和Fe3更高的激发能量,这表明Fe1团簇处于更高的氧化态(图2F)。通常,Fe团簇的低氧化态表明Fe-N键合可以共享更多离域的d电子,从而增强样品的电导率。
图3. 电催化ORR性能:(A)在O2饱和的0.5M H2SO4溶液中的ORR极化曲线,扫描速率为10mV/s,转速为1600rpm;(B)不同催化剂在0.75V电位下的电流密度和半波电位;(C)Tafel斜率;(D)不同转速下Fe2-N-C的LSV曲线;(E)Fe2-N-C的ORR稳定性测试,扫描速率为50 mV/s,电压范围为0.6-1.0 V;(F)Fe2-N-C在0.5M H2SO4含(红色)和不含(黑色)1M甲醇的LSV曲线。
LSV曲线显示,当Fex簇所含Fe原子数不同时,ORR活性依次为:Pt / C> Fe2-N-C> Fe3-N-C> Fe1-N-C> N-C。从半波电位和起始电位看,Fe2-N-C表现出更高的ORR活性(图3A)。
Fe2-N-C的半波电位(E1/2)为0.78V,在0.75V (vs RHE)下的比活性为16.4 mA cm-2(图3B),Tafel斜率为83mV/dec(图3C),由不同转速下的LSV曲线(图3D)得到电子转移数为3.9。Fe2-N-C在酸性ORR中表现出优异的循环稳定性,经20000次循环后E1/2仅负移20mV(图3E)。
此外,甲醇耐受测试表明,甲醇对Fe2-N-C在酸性条件下的ORR极化曲线几乎没有影响,这表明Fe2-N-C对甲醇具有优异的耐受性(图3F)。
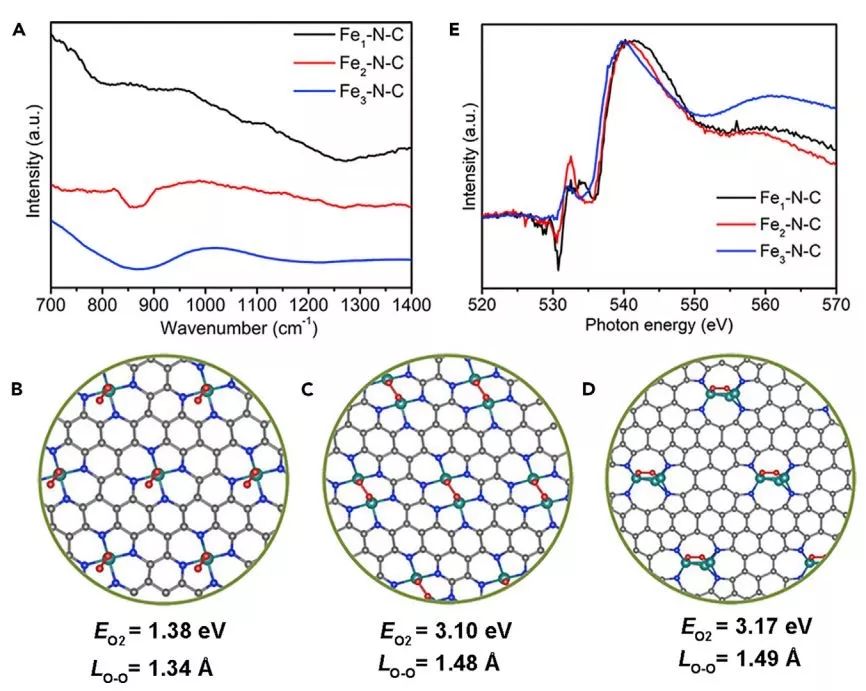
图4. O2吸附构型的表征和理论模拟:(A)在140 K下,在Fe1-N-C,Fe2-N-C和Fe3-N-C上吸附O2后的LT-FTIR光谱;Fe1-N-C,Fe2-N-C和Fe3-N-C上O2分子的最佳吸附构型,吸附能(EO2)和O-O键长(LO-O);(B)Fe1-N-C处的超氧吸附;(C)在Fe2-N-C下的过氧吸附;(D)在Fe3-N-C下的过氧吸附;(E)O K-edge NEXAFS光谱。
在LT-FTIR光谱上,位于1200-1350 cm-1和800-1000 cm-1的峰归因于超氧态和过氧态的O2物种的振动(图4A)。其中,Fe1-N-C主要表现出超氧态的振动,而Fe2-N-C和Fe3-N-C主要表现为过氧态的吸附,这表明Fe簇中的Fe原子数可以改变O2吸附构型。
理论计算结果表明(图3B-D),Fe1-N-C优先通过超氧态吸附O2分子,而Fe2-N-C和Fe3-N-C促进O2分子的过氧态吸附。
Fe2和Fe3位点有利于过氧态O2吸附,这很可能是因为Fe2和Fe3簇的原子距离比原子分散的Fe原子短。
O K-edge NEXAFS显示(图4E),532 eV的峰归因于与Fe 3d带相互作用的O 2p跃迁,这表明Fe2-N-C提供比Fe1-N-C和Fe3-N-C更高的跃迁强度。
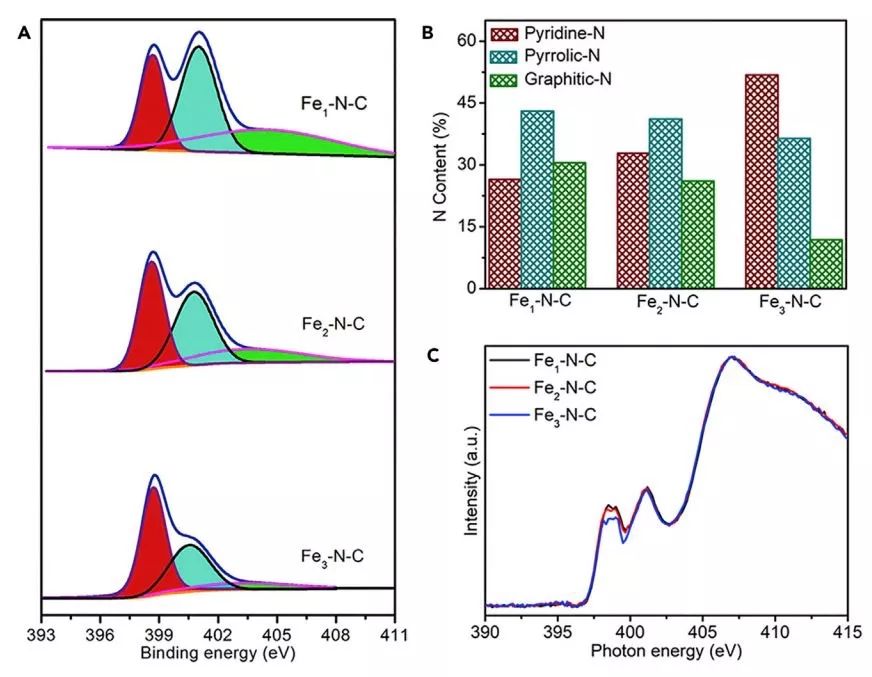 图5. 氮掺杂类型的表征:(A和B)N 1s XPS光谱(A)和吡啶N(深红色),吡咯N(深青色),石墨N(橄榄绿色)(B)的相应N含量;(C) N K-edge NEXAFS 光谱。
图5. 氮掺杂类型的表征:(A和B)N 1s XPS光谱(A)和吡啶N(深红色),吡咯N(深青色),石墨N(橄榄绿色)(B)的相应N含量;(C) N K-edge NEXAFS 光谱。
N物种(即吡啶N和石墨N)是决定ORR性能的一个重要因素。高分辨率N 1s XPS光谱可分为三个峰,位于398.6,400.7和403.8 eV 的峰分别归因于吡啶N,吡咯N和石墨N(图5A)。
随着团簇中Fe原子数的增加,吡咯N和石墨N的含量减少,而吡啶N含量增加(图5B)。N物种的这种变化可能由两个因素引起,首先,在热解过程中Fe团簇可以催化碳的石墨化,这决定了最终N物种的形成。其次,与Fe簇形成的Fe-N也会影响吡啶N的形成。
如N K-edge NEXAFS光谱所示(图5C),位于397.5-398.5 eV的峰对应于Fe1和Fe2促进吡啶N的跃迁,富含吡啶N的物质会增强ORR活性。
尽管Fe2-N-C和Fe3-N-C催化剂显示出类似的氧吸附模式,但Fe2-N-C可以在相同的Fe含量下为反应提供更多的催化位点。其中,石墨N和吡啶N促进了过氧态O2吸附,使得Fe2-N-C成为ORR最佳候选催化剂。
该工作在有机金属框架(MOF)孔道内负载了含有不同铁原子数目的Fe簇,在高温煅烧下实现了精准调控负载型Fe团簇原子数目,可以实现单原子Fe1、双原子Fe2以及三原子Fe3团簇结构。
随着团簇中原子数目的增加,可以实现氧气分子在催化中心的吸附方式由超氧态吸附向过氧态吸附方式转变;同时,含有不同数目的铁团簇在高温煅烧过程中可以调控氮的种类和含量。
该工作通过调控Fe团簇中的原子数目,进而调控氧气的吸附方式和衬底结构,最终实现了高活性、高稳定性的过渡金属氧还原催化剂,为酸性氧还原催化剂的设计提供了新思路。
Precisely Tuning the Number of Fe Atoms in Clusters on N-Doped Carbon toward Acidic Oxygen Reduction Reaction (Chem, 2019,DOI:https://doi.org/10.1016/j.chempr.2019.07.020)
https://www.cell.com/chem/fulltext/S2451-9294(19)30329-8?rss=yes.免责声明:本文资讯编译自学术期刊最新动态,以传播知识、有益学习和研究为宗旨,相关工作与本公众号无关, 如内容有误,请批评指正。
30W 底薪专业全职计算团队
120W 购买VASP商业版版权
吕梁云“天河二号”超算支持
您委托的计算服务商,专业吗?
深圳华算科技有限公司采用第一性原理计算与分子动力学、蒙特卡罗等方法相结合,对电池、催化、纳米材料等进行多尺度设计与模拟,专注为海内外催化、纳米及能源领域科研人员提供材料计算模拟整体技术解决方案。方向涉及材料结构、扩散、电导率、表面吸附能/吸附位、吸附分子构型优化、催化活性能、反应路径计算、OER、HER、ORR、自由能计算等。  长按识别下方二维码添加微信
长按识别下方二维码添加微信

 一步解决计算需求 为科研提速
一步解决计算需求 为科研提速










