Steffen Ventz, Brian M. Alexander, Giovanni Parmigiani, Richard D. Gelber, and Lorenzo Trippa
本刊负责人:马建辉; 审校:郝春芳; 翻译:钟晨菡
摘要
目的
:大部分肿瘤随机试验采用双臂研究设计评估新疗法与标准治疗的疗效。在这种设计中,较大比例的患者将被分配到对照组。相对而言,多臂研究设计采用多个试验组和一个共同的对照组进行比较。但是,传统多臂研究设计的主要难点是需要在试验开始时获得所有试验组的治疗措施(通常是来自不同公司的多种药物)。本研究的目的是评估一种平台研究设计(转臂设计)的潜在获益。这种设计可在滚动入组基础上增减试验臂数目。
方法:
我们在进行转臂设计时,以尽可能降低随机入组和试验数据分析的复杂性为目标。接着,我们对其在激素受体阳性、人表皮生长因子受体2阴性的进展期乳腺癌患者中的潜在优势进行评估。当下多个医药公司正在独立双臂研究中对来曲唑联合CDK4/6抑制剂的疗效进行研究。我们进行了一项模拟研究,来确定如下几项具体参数的减少对研究本身的效能的影响:样本容量、标准治疗患者数量、治疗平均起效时间(如果该方案已经在转臂试验中测试过)。
结果:
转臂设计含2-5个试验方案,和标准双臂研究相比,最多能够减少30%的样本总量。与五个检测特定治疗方式的独立试验相比,转臂设计的对照组入组患者个检测特殊治疗方式的独立试验入组患者的60%不到。而且,在某些实际情况下,部分治疗方案可以较双臂研究早15个月起效。
结论
:转臂设计适用于很多疾病的研究,而且,在实际情况下,其较标准双臂随机试验效率更高。
引言
药物研发昂贵且失败率高
1
。双臂研究设计常被用于评估新疗法与标准治疗的疗效。所以,每个试验药物均需开展一个随机试验。与双臂研究相比,多臂研究设计具有多个试验组和一个共同的对照组
2,3
。如果每个试验药物均与相同对照药物进行独立的两臂试验,那么,重复设置了多个对照组,降低了研究效率。此外,与双臂研究相比,多臂研究的成本更低
2,4
。但是,只有一些小部分临床试验是多臂研究。多臂试验要求同步评估所有的治疗措施,但是,这些药物很难在药物研发的相同阶段同时出现。
最近,可以通过增加或减少试验组解决上述不足的平台试验设计引起关注
5-10
。I-SPY II,
11
STAMPEDE,
12
AML15和AML16,
13,14
神经学的NET-PD
15
试验以及精神病的CATIE
16
试验都采用了上述方法。I-SPY II,
5,6,11
运用了Bayesian结局适应性设计,是该平台试验在肿瘤学研究的一个应用。运用结局适应性随机算法,根据患者的分子表型,将患者分配进入不同治疗组。Bayesian算法能够将该试验不断累积的数据转化为随机分组的概率。国家癌症中心的MATCH
7
试验计划运用篮子设计增加额外的试验组。在这种情况下,每个试验臂可形成一个由在生物标志物界定的亚组人群。
最近,Saville和Berry
17
研究了一种用新试验组取代了多臂研究中原有试验组的Bayesian设计,这种研究思路早有论述
15,18,19
。平台试验设计尚处于早期阶段
9
,实例还不多
5-7,12
。协作组和癌症中心对高效筛选试验药物的需求促使我们开展本次研究。
我们构思了一种平台试验设计。这种设计从多个独立的试验(每个试验测试一个或少数几个临床假设)转变为一个更高效的、一体化的药物筛选过程。我们在与标准多臂试验相比较的基础上增加最小复杂性来阐明平台实验设计的获益。
我们要介绍一种特定的研究设计--转臂设计。转臂设计是对已有的多臂研究的拓展
20-
22
。
虽然我们认识到Bayesian研究和适应性研究的好处
2
3-27
, 更简单的研究设计仍有可能应用于实践且有可能加快其更广的应用。与标准随机分组的多臂试验相比,转臂设计不需要其他额外的统计专家。通过标准方法就可以计算出规定的效能和I类错误上的样本容量并不需要特定的用于反应适应性研究设计的模拟软件辅助,其样本容量就可在规定的效能和I类错误上得出的。该研究可以用于不同的研究终点,包括对治疗的二分类结局事件和II期探索性时间事件终点或者III期验证性研究。这里,我们主要关注的是时间事件终点。
虽然在临床研究过程中增加试验组是可以接受的,这里,我们在国际乳腺癌研究会的背景下对这种转臂设计在进展期乳腺癌患者的研究进行分析和评估进行。我们关注PI3K抑制剂在绝经后、激素受体(HR)阳性和人表皮生长因子受体2(HER2)阴性进展期乳腺癌患者的应用。若干试验正在探索某些药物(帕博昔布, LY2835219
,
LEE011
,
BYL719和依维莫司)与标准的内分泌治疗(来曲唑)联用与那些双臂对照研究的疗效对比。我们进行了一项模拟研究,来确定如下几项具体减少的量:样本容量、标准治疗患者数量、该治疗平均起效时间(如果该方案已经在转臂试验中测试过)。
方法
转臂设计, 一个用于增加和删减治疗组的设计平台。文中的转臂设计(图1)刚开始是标准的双臂或多臂研究。每个试验组入组的病人数量n是最大入组量。患者具有相等的概率进入试验组或对照组,如图1B所示。样本容量大小n是根据特定的I/II类错误率得出的。
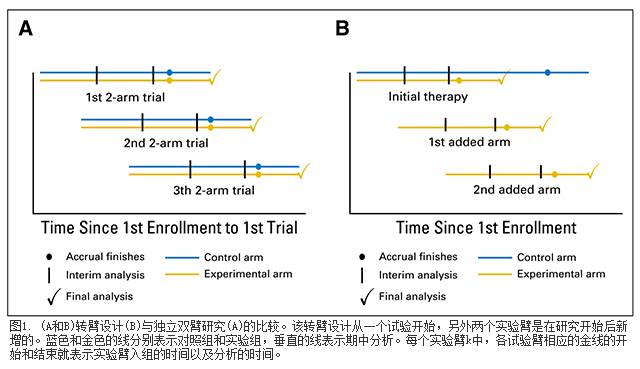
试验臂的最大数量是特定的(模拟研究中最多4个),可以添加到转臂研究中(图1)。若有新的、可获得的、合适的治疗方案,可增加该试验臂。此时,样本量需要重新计算(数据见附件), 所有病人都以同样的可能性进入原始试验组、对照组和额外增加的试验组。每个试验组入组的病人数量都是其最大数量。各组患者可根据其特点进行分层,详见数据见附件。
图1示转臂研究。金线表示的是每个试验臂k和治疗k最终行分析之间的时间间隔。图1两组队列可见随着时间的变化,实际进行的实验臂数量也在不断变化。最初,只有一个试验组招募患者,然而之后,有三个试验组同时招募患者。
如果试验臂k没有很早结束,那么在持续招募行k治疗的患者过程中,那么n个患者同时随机分配进入对照组(图1)。由于对照组会在所有试验臂(最初的和后期添加的,或是招募了n名患者或在中期分析时已经停止)结束后停止。
我们把时间事件作为主要研究终点。主要对疗效进行分析,单侧零假设Hk:假设实验组与对照组的危险比(HRk)≥1。其他研究终点和备择假设可以很快根据上述变化进行调整。实验组k的临时和最终分析的结果与双臂研究常用的分析一致。若期中分析时试验臂k结果提示无价值或疗效不佳可提前终止(J表示各试验臂期中和最终分析结果,分别对应j = 1
,
...
,
J-1)。所有与治疗k相关的对比分析只对k臂招募患者期间随机进入对照组的患者的结局进行,上述患者只占整个研究的一小部分(图1)。对于k臂来说,在log-rank数据分析基础上,在k臂开始招募患者后(nj
,
j = 1, ..., J),在k臂或者对照臂患者中观察到某事件时对标准治疗组进行连续分析。通过对k臂开始招募患者至j-th期中分析期间k臂治疗患者的结局和随机分配进入对照组的患者的结局,对k臂在j-th期中分析时进行t检验(经仔细观察和检查)。
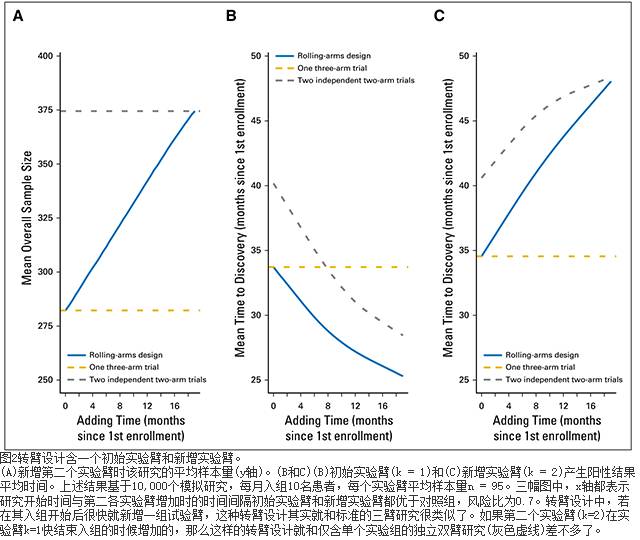
图2中显示的是一个实验臂自入组第一个病人开始加到另一个初始为双臂研究后x = 1
,
2
,
...
,
或20月后的转臂研究的例子。两个实验组的生存曲线都是指数式的,其中位时间为15月,对照组为10月(n=95)。图2A表示的是加入第二个实验组后不同时间点(x轴)转臂设计平均总体样本大小(y轴)。图2B 显示的是实验臂k=1在加入第二个实验臂后随时间点(x轴)变化的平均疗效评估(y轴)。相似的,图2C显示的是实验臂2的平均疗效评估时间。
模拟研究
内分泌治疗是HR阳性、HER2阴性乳癌女性患者的标准治疗
28
。转移性乳腺癌不可治愈,最终内分泌治疗也会出现耐受。包括辉瑞、诺华和礼来在内的多家医药公司,其选择性CDK4/6抑制剂以及哺乳动物雷帕霉素靶蛋白抑制剂的研发已经进入最后阶段。现在,已经有若干为研究CDK4/6抑制剂(瑞博西尼,temsirolimus
,
帕博西尼, LY2835219和BYL719)与内分泌药物联用在HR阳性、HER2阴性的绝经后转移性乳腺癌患者中疗效的II期和III期双臂研究(如,NCT00721409
,
NCT02246621
,
NCT02107703
,
NCT01958021
,
NCT01958021
,
和NCT00863655)。
我们还运用转臂设计为某项旨在研究来曲唑与5种抑制剂(瑞博西尼
,
temsirolimus, 帕博西尼,LY2835219和BYL719)联用疗效的研究开展了模拟研究。我们假设为对照组中的患者接受来曲唑和安慰剂的治疗。入组标准为绝经后HR阳性、HER2阴性的转移性乳癌患者。无论是要入组或出组,在最初组、额外增加组还是仍在研究中的人群其入选和排除标准一致。
首要研究终点是无进展生存(PFS),从随机入组开始至影像学可见疾病进展或患者死亡。在来曲唑与安慰剂联用组,中位PFS是10个月
29,30,
也可使用诸如总体生存l(OS)或疗效等其他观察指标作为研究终点。只需要满足其和预设转臂设计有微小的差异即可。我们用PFS来保证我们的模拟研究与前述双臂研究一致。若疗效假设与平台研究或者双臂设计的研究一起测试,应选择像PFS
,
OS
,
或者疗效作为首要研究终点,这在临床中是至关重要的。
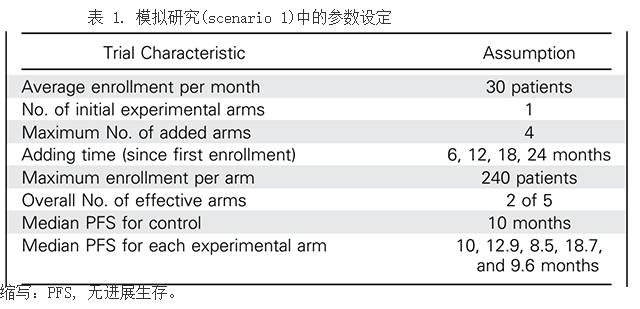
表1对模拟研究的假设进行了总结。本研究开始时为双臂,计划随访时间为22月。样本容量是在单侧I类错误误差范围为2.5%的概率为80%、HR为0.75的条件下计算所得。我们对来曲唑所得数据用威布尔概率模型分布进行拟合
29,30
。
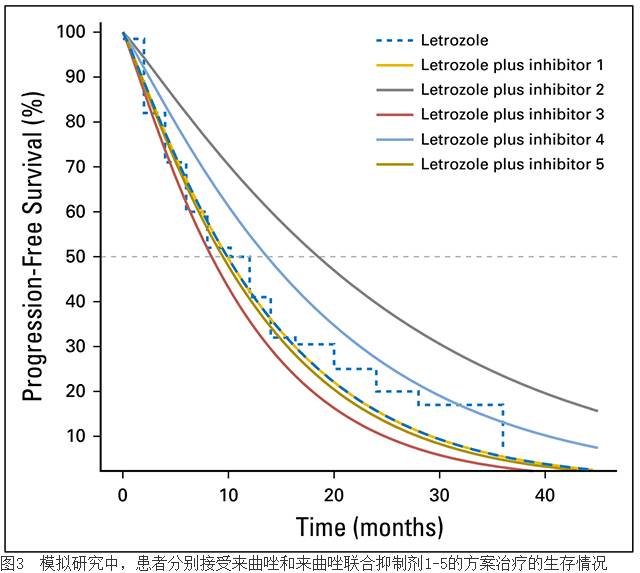
中位PFS为10月。其余四种方案分别在试验入组第一名患者后6,12,18和24月后加入本研究(图3)。我们建立了一种比例危险率模型(HR分别为1,0.75,1.2,0.5,和1.05)。最近的PALOMA-1研究
30
中,其HR为0.5,提示对PFS明显延长。
每个实验臂最大入组患者数量为240人。对每个试验臂k=2
,
...
,
6,来说,随机分配进入k臂或者控制臂在k臂开始之后,分别在观察到nj = 96
,
192
,
288以及383例PFS事件之后,行期中分析和最终分析。期中分析以O’Brien-Fleming效能分析法
20
和Jennison-Turnbull无效指数计算法
21
为基础。在模拟研究中,如果转臂研究终止较早,比如,所有试验组都因无效而停止,然后,在一种新的治疗可行,一种新的转臂研究就开始了。额外的实验臂可以在这之后加到第二个转臂研究中。
我们将转臂设计与独立的双臂研究相对比。在5个双臂研究中,总体入组率与转臂设计的入组率相同。我们也考虑了在转臂设计中使用Bayesian适应性设计。实验臂被当作是转臂设计并加入研究中。另外,使用可获得的提示治疗影响的Bayesian设计通过适应性地随机分配患者。Bayesian版本的转臂设计可以增加随机进入最有前景的试验性治疗方案的可能性,但其具体人数还未确定。具体研究设计详见数据附件。
结果
表2和图4对模拟研究的主要结果进行了总结。图4的前两张图分别是转臂设计(图4A) 或五个独立的双臂研究(图4B)在10,000个模拟研究中每个试验臂和对照臂平均入组量。图4C通过运用不同研究设计对比接受标准治疗的患者数量。
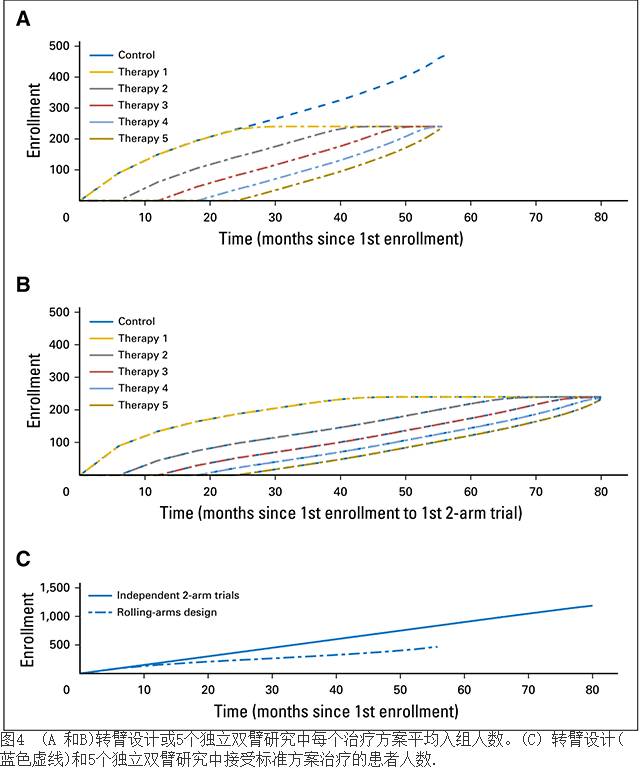
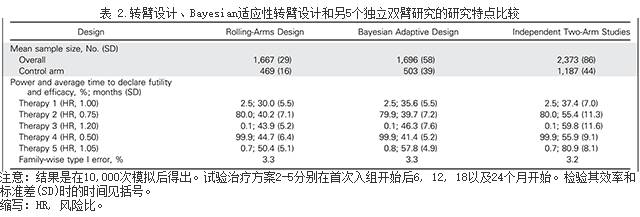
转臂设计的样本大小约为其他5个双臂研究样本量之和的三分之二(1,667 v 2,373患者)。一般来讲,28.1%的患者进入对照组,而与之相比,双臂研究时则对照组人数占50%。如果5个实验治疗组能同时启动,那么这个六臂研究的总体平均每臂入组1435名患者。
在我们的模拟研究中, 总的来说,Bayesian设计平台对筛查实验臂的样本需求对每个实验臂的样本量需求较独立的双臂研究少三分之一左右(1,696 v 2,373 患者)。这和转臂设计相似。而Bayesian设计平台的总体平均样本量稍大(1,696 v 1,667 患者)。
评估有效性和无用性的时机
转臂设计和独立双臂设计有相近的I类错误率和效能(表2)。此外,在我们的模拟研究中,转臂设计和独立双臂的总体I类错误率几乎一致(3.2%)。对两个两种设计来说,实验治疗组3和5的I类错误均小于2.5%,其HR分别为1.2和0.5。
转臂设计及5个独立双臂研究评估治疗有效性和无效性的时机相差很大。例如,对于治疗试验3来讲,其中位PFS延长了2.9月,有统计学意义,转臂设计需随访40.2月(标准差
,
7.1月),而独立双臂研究需随访55.4月(标准差
,
11.3月)。本文中所述平均评估时机指的是从转臂研究开始直至能够获得统计学意义的时间,此时,某种治疗的零假设Hk被拒绝,而且,转臂设计中止了三个无效试验臂,和独立双臂研究相比,分别平均要早7.4
,
15.9和30.5月。
Bayesian设计平台的效能和I类错误率与其他两种代替性研究相近,但其平均评估治疗的时机比转臂设计和独立双臂的更短。对于某些有效的试验组来说,从开始入组开始平均需要39.7和41.4月,而独立双臂研究则需要55.4和55.9月,转臂设计则需要40.2和44.7月。在模拟研究中,有三组无效试验臂(无统计学意义),比双臂独立样本研究所需时间平均短1.8
,
13.5和23.1月,但比转臂设计中所结果分别平均晚5.6,2.4和7.4月。与非适应性转臂设计相比,Bayesian设计在疗效评估时间上的获益更大,因为适应性随机分组可能会导致有效的实验臂的随机分组可能性增加。
模拟研究的变量
常规转臂设计和Bayesian版的转臂设计效率较独立双臂研究明显提高。现在,我们想通过改变模拟研究的某些参数,如中位生存时间、有效臂与无效臂的顺序、累计率和新增实验臂的数量,并观察它们改变后该研究的敏感性(表3)。因为转臂设计较Bayesian设计,更易在医生中应用和开展,因此我们重点关注转臂设计。

我们用和之前相同的假说、不同的有效臂与无效臂顺序以及实验臂总数,重复模拟研究。我们从如下实验臂中挑选了三个额外的实验组;前三组实验臂是无效的,后两组是有效的(详见数据附件)。转臂设计的获益和其他研究的获益相似。转臂设计的的平均样本量比独立双臂研究的样本量少约30% (患者人数:1,666-1,670 v 2,369-2,394人)。而且,转臂设计的对照组比独立双臂研究中的对照组所需患者更少(患者人数:470-472 v 1
,
184-1,197人)。
接着我们加快月入组率,从30名患者每月增至40名患者每月和50名患者每月(详见数据附件)。更快的入组率使得转臂设计总体样本量由1,667人增至1,765人(40人/月)和1,882名患者(50人/月)。与独立独立双臂研究相比较而言,样本数量只是从2,373名患者增加至2,392和2,398名患者。这种差异是双臂研究中因运用提前中止原则所致。
更快的入组率也会缩短实验臂评估疗效的时间,例如,转臂设计中实验臂2的评估时间由40.2月缩短至32.3月和27.4月,双臂研究中实验臂2则从55.4月缩短至42.7月和34.6月。相似地,转臂设计中实验臂4(HR,0.5)的平均评估时间由44.7月缩短至38.7月和35.3月。
在入组率为40名患者每月和50名患者每月的情况下,独立双臂试验较转臂设计平均晚7.8月和5.7月才能进行评估。在无效臂中的结果也相似。转臂设计平均要早4至12月进行无效性评估。这里平均评估时间指的是模拟研究间的平均评估时间,那时无效臂或是因其无效而提前中止,或是已经获益且不拒绝Hk。
接着,我们又开展了另一项研究,分别增加一个、两个、三个或四个实验臂,每隔6月增加一个实验臂(详见数据附件)。以仅在试验开始后增加一个实验臂的研究为例,该实验臂的增加会导致转臂设计的平均总体样本量变成809名患者,其中有329名(40%)患者进入对照组,而独立双臂研究的平均总体样本量为957名患者,其中有479名(50%)患者进入对照组.
人群流动和非同期对照增大了I类错误和估计偏倚
我们对转臂设计中几个随着时间变化的参数进行分析。根据对照组中线性增加的不同中位PFS将病人分为0.5
,
1.0
,
1.5或2.0月(每隔10月),我们发现患者的组成会随着时间变化。我们在模拟研究中也看到了这种趋势(包括随着时间变化个人PFS的分布也发生变化)。我们在实验臂中运用了相同的时间变化比例因子并像之前模拟研究中那样将实验臂与对照组的HR分别稳定于1.0
,
0.75
,
1.2, 0.5和1.05。
为了进行比较,我们采用另一种设计和数据分析方法,该方法与转臂设计几乎相同,但其会将实验臂的数据与所有随机分配进入对照组的患者数据(包括实验臂开始前随机入组的患者)进行比较。
数据附件中可见实验臂3和5的I类错误以及它们在10,000个模拟研究(存在患者人群漂变)之间平均HR。为了对HR进行计算,我们为每个模拟研究建立了Cox比例风险回归模型。对于在试验开始后18月和24月增加的实验臂3和实验臂5来说
,
其I类错误率可能因人群流动而增大(若研究早期随机进入对照组的患者也纳入分析时)。以实验臂3为例,若试验期间对照组的中位PFS每隔10个月改变1.5个月,其I类错误率会增加至4%(原计划的I类错误率为2.5%)。仅使用同期入组的对照组数据的转臂研究的I类错误率似乎并不受这种时间趋势的影响,其所得HR偏倚更小。通过比较可知,将实验臂开始前就进入对照组的患者也纳入分析会使得结果产生偏倚。在时间趋势较弱的实验臂5,其平均HR为0.91(真实的HR是1.05) 。
模型误设
我们通过非比例风险模型对转臂研究中涉及参数的稳定性进行评估。我们对标准治疗数据用对数-logistics加速失败时间模型
31
。接着,我们在各实验组保持对数-logistics模型不变
,
挑选出与之前模拟研究的结局数据中用过的威布尔概率模型相匹配的中位PFS时间(见数据附件)。运用风险比例模型中获得的模拟研究的结果与工作特性几乎一致。
讨论
药物研发主要关注生物学通路,临床试验主要是为了发现标记物亚组人群。鉴于试验入组人群累积量增长较慢,研究的设计应该提高新实验组评估的效率。新兴靶向治疗所处研究阶段不尽相同。传统的多臂研究设计仅适用于试验开始时所有实验组可实施的情况下。平台试验允许研究者增加或减少试验臂,进而加快了筛选试验方案的过程。
我们对转臂研究进行了讨论,它代替Bayesian适应性设计,更易进行实施,更为生物统计学家、研究者及患者所接受。与设计和开展新的单一试验方案的研究相比,转臂设计大大提高了试验的效率。
在对结果进行分析时,我们借鉴了Freidlin等
3










