正文
传统的计算机辅助药物设计一般是基于药物和靶标之间的亲和力算法的优化,然而药效的作用不仅仅和平衡态的亲和力有关,同时与靶标蛋白之间的滞留时间相关。因此,高效的药物设计不应仅仅关注平衡态的亲和力分析,同时需要在传统的设计策略的基础上加入药物在靶点中“寿命”的分析。虽然实验技术也可以进行滞留时间的测量(koff),但除了量化滞留时间以外,了解分子解离的决定因素以及解离的途径和能量壁垒也同样重要。这些测定包括但不限于:关键的蛋白-药物和蛋白-蛋白联系,蛋白的构象状态,配体柔性以及溶剂影响等等。
c-Src激酶是一种非受体细胞质酪氨酸激酶,参与多种细胞信号传导活动,其活性升高与多种癌症(包括结肠癌,肝癌,肺癌和胰腺癌)的发展有关。达沙替尼是美国食品和药物管理局批准的药物,用于通过阻断c-Src以及Abl激酶来治疗慢性骨髓性白血病和其他相关疾病的患者。使用全原子模拟来了解达沙替尼如何解离的详细过程可能对设计可控制滞留时间的药物具有重要的意义。
哥伦比亚大学的B. J. Berne教授在《SCIENCE ADVANCES》发表了题为“How and when does an anticancer drug leave its binding site?”的文章,他们通过元动力学( metadynamics)方法对抗癌药物达沙替尼和c-Src激酶的结合态进行了增强采样动力学模拟,成功的获得了配体药物和蛋白之间的解离过程的轨迹。作者能够计算出与实验测量结果一致的药物滞留时间,并确定与达沙替尼解除结合速率步骤有关的机制特征,这些步骤也与实验相符。这种原子级分辨率的视角揭示了以前无法解释的实验观察结果,例如为什么针对某些c-Src激酶构象形式的靶向工程可能无益于增加药物的滞留时间。B. J. Berne教授的工作还提供了直接的证据,表明水在药物解离过程中不仅是作为溶剂的旁观者,还是一个非常积极的参与者。作者的研究结果标志着在统计可靠性的情况下计算药物未结合途径和速率的能力向前迈出了重要一步,并从多疾病靶点c-Src激酶中获得了对药物解离的复杂性质的理论信息。
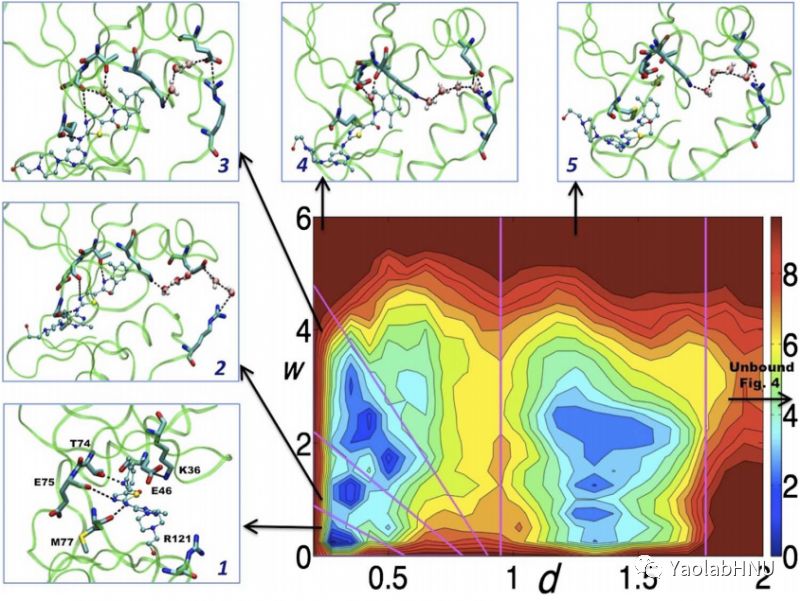
图1右下角大图为自由能景观图(free energy surface,FES),横坐标d为配体与结合口袋之间的距离,纵坐标w为结合口袋的水化态,1-5的小图为对应的亚稳态的中间体,关键残基,和达沙替尼与c-Src激酶之间相关的氨基酸残基。能量单位为kcal/mol,小分子配体用球棍模型所表示。该图展示了整个解离过程的关键亚中间态。
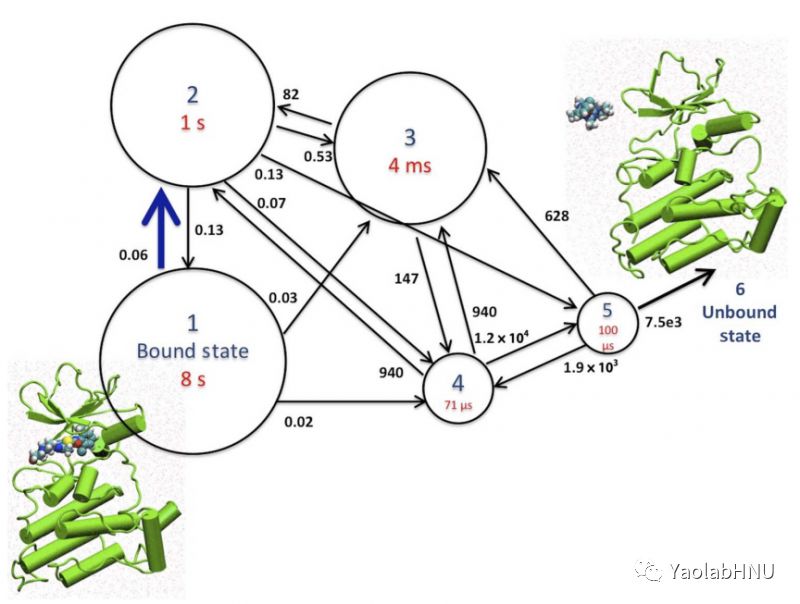
图2表示的为达沙替尼从结合态到解离态之间的变化网络,速率常数为s-1。 有关各种状态的定义,请参见图1。 各种状态的圆的半径与其各自的停留时间近似对数成比例。

图3表示多个蛋白结构的比对,展示了αC螺旋和谷氨酸(PDB 3G5D中Glu46)的旋转。橘黄色表示平衡后αC螺旋朝内的结构,红色表示αC螺旋朝外的结构,红色部分对应于图1的state3,为作者模拟所获得的结构。蓝色表示的为从PDB数据库(PDB 4YBK)获得的αC螺旋朝外的结构。可以发现计算与实验结果吻合度非常高。同时可以看出Arg121和Glu46之间稳定的联系,同时与图中紫色表示的活化环之间有相互作用。
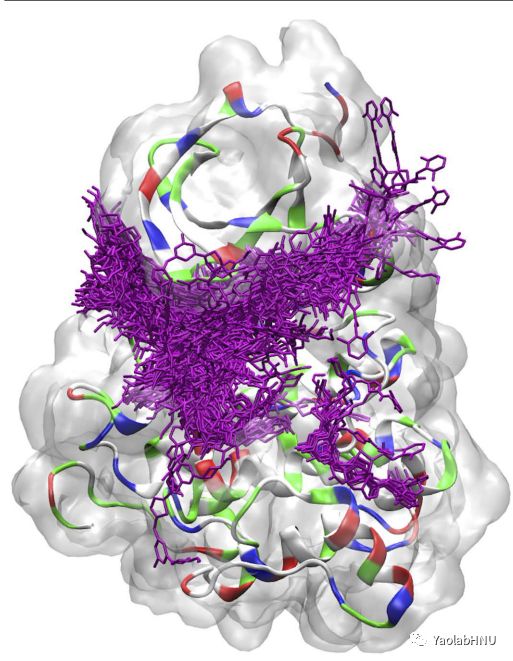
图4表示达沙替尼从主结合口袋脱离的快照(即d>1.8nm;参见图1),可以清楚的发现c-Src激酶表面存在着相互吸引的热点。来自满足d> 1.8nm的未结合轨迹的各种达沙替尼快照在蛋白质上被标记为紫色。
该研究描述了c-Src激酶和小分子抗癌药物达沙替尼之间解离的动态过程,并且获得了和实验数据一致的药物滞留时间。同时作者发现结合口袋和配体之间发生了水合作用,这给对提高药物滞留时间的药物工程提供了较大的理论依据,即不应该仅仅增加配体-结合口袋之间的联系,同时需要关注如何控制水进入结合口袋。同时作者发现了αC螺旋介导的保守Lys-Glu盐桥作为解离的分子开关,这一理解具有明显的药理学意义。最后作者也指出了现在该方向的一些瓶颈,最大的瓶颈即计算花费过于庞大,并不能计算很长的滞留时间(截至文章发布前已有计算11min滞留时间的文章发表1),过大的花费也限制了其应用。在准确的基础上减少计算时间可能是未来该领域急需解决的问题之一。
Pratyush Tiwary, Jagannath Mondal and B. J. Berne, How and when does an anti-cancer drug leave its binding site, Science Advances, 3, e17000,1-8 (2017) DOI:10.1126/sciadv.1700014
参考资料: [1] Unbiased Molecular Dynamics of 11 min Timescale Drug Unbinding Reveals Transition State Stabilizing Interactions,J. Am. Chem.Soc. 2018, 140, 618−628 DOI:10.1021/jacs.7b08572

Bruce J. Berne,男,博士,美国哥伦比亚大学教授。他于1961年在布鲁克林的纽约市立大学获得化学学士学位,并于1964年获得芝加哥大学化学物理学博士学位。 他早期的主要贡献是展示如何使用记忆函数模型进行非马尔可夫动力学的液体模拟。 抵达哥伦比亚后不久,他和他的学生GeorgeHarp开发了方法论并进行了第一次有关分子液体的分子动力学模拟。他发表了近350篇出版物和四本书,为时间相关和记忆函数理论,光散射理论,经典和量子蒙特卡罗方法以及分子动力学,反应速率理论,量子液体理论,小规模和大规模的疏水效应,水分子的可极化力场的设计,以及最近蛋白质的生物物理学,特别是蛋白质 -配体结合理论等提供了重要的贡献。
作者主页
http://www.columbia.edu/cu/chemistry/groups/berne/berne.html
编辑:KWY
审核:芳芳是老虎
推送:Kumquat Lemon





