听取患者反馈和查阅三期临床试验数据后,评估新药安全性与有效性的FDA咨询委员会匿名投了全票,支持美国Spark Therapeutics公司的基因疗法“Luxturna”(针对RPE65基因异常的患者)上市。能否改变莱伯先天性黑蒙症无药可医的局面,只差临门一脚:2018年1月前,FDA将做最后决定。

本文转载自“澎湃新闻”,作者“王盈颖”。
对莱伯先天性黑蒙症患者而言,失明是一经出生就注定,需要一生去逐渐接受的事实。
它是一种在新生儿出生一年内就发病的罕见遗传病,每10万人中有2-3位不幸罹患。捕捉疾病痕迹的场景往往发生在,婴儿的目光不会追随悬挂晃动着的玩偶,对刺眼的光源却毫不避让。
随着年岁增长,患者的视野会是轮廓和阴影的集合,并在中年时,彻底跌入漆黑的失明深渊。而且,无药可医。
对10月中旬,在美国食品药品监管局(FDA)咨询委员会面前做术后陈述的几位莱伯先天性黑蒙症患者来说,他们大多不会后悔自己选择接受了一个基因疗法的临床试验,来对抗失明的到临。
12:0——听取患者反馈和查阅三期临床试验数据后,评估新药安全性与有效性的FDA咨询委员会匿名投了全票,支持美国Spark Therapeutics公司的基因疗法“Luxturna”(针对RPE65基因异常的患者)上市。能否改变莱伯先天性黑蒙症无药可医的局面,只差临门一脚:2018年1月前,FDA将做最后决定。

Carper姐弟俩都患有莱伯先天性黑蒙症,他们都接受了基因治疗的临床试验。来源:《金融时报》
若成功上市,这将是美国第一款针对遗传病的正式基因疗法,虽然2016年9月美国FDA快速通道在有争议的情况下有限度地批准了美国Sarepta公司治疗杜氏肌营养不良的基因治疗药物Exondys 51。在全球范围内,第一款矫正遗传病的基因疗法是荷兰UniQure公司2012年在欧洲上市的Glybera。
作为用于治疗罕见病脂蛋白酯酶缺乏症的基因治疗药物,Glybera赚得名声,但落得一个令人唏嘘的结局。4月,UniQure公司宣布,不再重新申请Glybera的销售许可,10月后将退出市场。在100万美元/次的收费下,Glybera上市以来仅出售一次。
同样预测双眼治疗价格为100万美元的Luxturna能否带来一个新的开局?这尚在人们谨慎的期望之中。
但就基因疗法在莱伯先天性黑蒙症上的应用而言,用浙江大学医学部基础医学系教授、发现莱伯先天性黑蒙症第18个致病基因的祁鸣的话来说,这是“非常震撼的”。

祁鸣
1RPE65
1869年,DNA首次被分离出来,元素周期表被发现。就在这一年,德国眼科医生西奥德·莱伯(Theodor Leber)首次描述了发生在婴儿时期,因遗传性视网膜疾病而造成的严重视力损伤。莱伯先天性黑蒙症(Leber Congenital Amaurosis,LCA)由此得名,开始进入历史的视野。
在解释这种疾病为何造成视力困难前,先要提到维生素A在“看见”中扮演的角色。
早在古埃及时期,人们已经开始模糊地意识到,糟糕的摄食条件和夜盲症之间的关系。直到第一次世界大战期间,研究人员才进一步缩小范围,将食物中的维生素A摄入不足和营养性夜盲症联系起来。
20世纪30年代,美国科学家乔治·沃尔德(George Wald)首次确认,维生素A在视觉系统中扮演着何种角色。他发现,视网膜中的感光色素——视紫红质是由一种蛋白和维生素A组成的混合物。进一步地,沃尔德提了“视循环(Visual Cycle)”的概念。
这一循环是什么过程呢?第一步,视紫红质接收到光线后,会改变维生素A的性状,将其与和蛋白分离,产生能量,将光能转化为电能,以此将信号沿视神经传导到脑区。之后,原先的维生素A会被回收利用,恢复之前的形状,和蛋白重新结合,如此不断往复。正因为在视网膜上的发现,沃尔德获得了1967年的诺贝尔生理学或医学奖。
RPE65基因被发现是很多年后。1993年,美国眼科研究所的研究员迈克·雷蒙(T.Michael Redmond)发现,在视循环中,一个蛋白在合成维生素A中必不可少,而这种蛋白正由RPE65基因编码产生。
1997年,RPE65基因被确认与莱伯先天性黑蒙症相关。RPE65基因“罢工”的患者会导致维生素A无法回收,不能和蛋白再度结合,从而导致视紫红质缺失,视觉信号无法传导至大脑,自然也就无法看清。
RPE65由此成为莱伯先天性黑蒙症第二个被发现的致病基因,所以该基因出现问题的患者也被称为LCA2型患者。LCA2型是至今发现的22个致病基因中最常见的类型之一,约占总患者的6%。
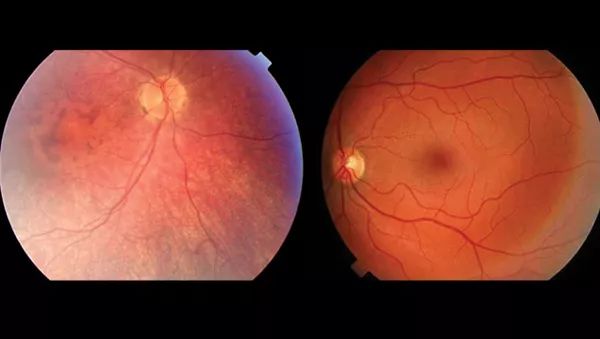
左为LCA患者,右为健康人。两者的视网膜对比,LCA患者的血管组织更少,视网膜更薄,黄斑有所变化,没有中央暗斑。
LCA2型患者发病机制相对简单,“零部件”完整无缺,但需要一个能正常工作的RPE65基因来“重启”中断的视循环。
Spark Therapeutics公司经过三期临床试验,或即将上市的基因疗法“Luxturna”正是给LCA2型患者补上了正常的RPE65基因。但,用什么来输送这一基因?如何让这一基因“扎根”在患者体内?
2“运输车”病毒
对。RPE65基因是“搭乘”病毒这辆听起来有些可怕的“车”进入患者体内的。
这并不是Spark Therapeutics公司的首创,事实上,在1990年,人类历史上第一次基因治疗就已经用病毒作为运输工具,将目标基因引入患者体内。
比起针、电击、脂质颗粒等微量DNA运输工具,病毒被看作是自然界的基因运输高手。病毒的身躯很小,结构很简单,所“内置”的遗传信息也不多。但病毒的强大寄生能力让它的威力大大增加——病毒可将自己的遗传信息嵌入到宿主细胞中,代由宿主细胞来帮助自己繁衍后代。
这一特性被科学家所利用。如果病毒经过改良,不再具有致病性,阻止其在体内的复制,同时又让病毒携带着目标基因,是不是就可以达到基因引入的目的呢?
在1990年那场具有里程碑意义的手术上,美国国家卫生院的威廉·安德森(William Anderson)在重症联合免疫缺陷病患者身上测试了这一大胆设想。安德森选择的病毒是莫罗尼小鼠白血病病毒,经过改造,病毒能定位和进入人类细胞,但无法生成新的病毒。
这些经过改良的病毒携带着重症联合免疫缺陷病患者所欠缺的正常腺苷脱氨酶基因,顺利地为患者补上了,原本因丧失免疫功能而只能生活在封闭气泡舱里的患者终于得以像正常人一样生活。
但将普通病毒作为“运输车”的无方向盘“恶魔性”很快显现出现。1999年,英国和法国的医生“改进”安德森的方法,将携带所需基因的放进了造血干细胞。预期要治疗的疾病是治好了,但却造成多位患者都得了白血病。
问题出在,病毒将所需基因运输到了人体基因组DNA的什么位置?各司其职的DNA片段会因为突如其来的插入者而造成功能损害。
祁鸣的比喻很生动。他对澎湃新闻解释,“就像天上掉一个东西下来,那么它必须要有一个机场,或者是大海、沙漠,才能够安全着陆。不能掉在杭州,也不能掉北京,不然把我们人砸死掉了对不对?”
而1965年被科学家所发现的腺相关病毒(AAV)是一个更为理想的“运输车”。AAV是目前所发现的结构最简单的单链DNA缺陷型病毒,在1984年被科学家提出,可为人体细胞引入外源DNA。
“AAV的特点是能安全地着陆在某一个地方——第19号染色体,正好那地方相当于大海或沙漠,是没有基因的,无人居住。这一伟大发现是上世纪80年代末,美国匹兹堡大学中国留学生、我的同班同学肖啸博士的论文成果。”祁鸣说。正因为“着落点”是无人区,鲜少对已有DNA片段产生作用,AAV病毒作为“运输车”要更为安全。
而Spark Therapeutics公司的“Luxturna”正是通过AAV病毒,将RPE65基因安全“着落”于视网膜。
3布里犬“Lancelot”
明晰了LCA2患者的病理机制,找到了可能的解决方法,剩下要做的:证明方法安全、有效。
而在人体上做试验,即临床试验之前,科学家必须先在动物模型上验证方法的有效性、安全性。
正在科学家们埋头于实验室,想通过常规的方法——敲除RPE65基因,构建LCA2动物模型时,大自然给了科学家一个馈赠:一只天生失明、长着浅棕色长毛的布里犬。当兽医发现这只布里犬时,它的症状和莱伯先天性黑蒙症患者类似,后经检查证实,它确是RPE65基因出现故障!
一个“完美”的动物模型从天而降。“Luxturna”基因疗法的核心人物:美国宾夕法尼亚大学眼科教授琼·贝内特(Jean Bennett)和同为眼科教授、她的丈夫艾伯特M·马奎尔(Albert M. Maguire),也在此时开始了他们和基因治疗莱伯先天性黑蒙症长达10多年的接触。

琼·贝内特(Jean Bennett)
2001年,一篇以贝内特为通讯作者、马圭尔为参与者的论文在《自然-遗传学》上发表。在那只类莱伯先天性黑蒙症的布里犬上,贝内特和合作者们测试了用AAV病毒携带野生型RPE65基因进行治疗的有效性。
测试结果是:布里犬的视力得到显著改善。治疗后数周,布里犬不再撞倒路上的障碍物,可以顺利地自我导航,通过障碍路段。
这只不幸又有幸的布里犬被取名为“Lancelot”,和神话中勇敢、乐于助人的圆桌骑士同名。
同年,为纪念它所作贡献,Lancelot被带到美国国会大厦前,留下了“英雄照”。

Lancelot的“英雄照
有依可循的是,至少在术后8年,Lancelot依然享受着基因疗法带来它的改变。
人体临床试验
Lancelot身上的科学奇迹没有顺理成章地推动人体临床试验的开展。这和基因治疗陷入低谷的大背景有关。
1999年,美国18岁男孩杰西·基辛格(JesseGelsinger)在一项基因治疗的临床试验中不幸去世,致死原因是免疫反应带来的细胞因子风暴。加之前文所述,2003年,5位接受基因治疗的儿童不约而同地患上白血病。一时之间,基因治疗从之前遍地开花的狂热,一下子跌到人人避之的谷底。
各国监管机构亮起了红灯,出台了严格的限制措施,和基因治疗相关的科研经费一度“断粮”。
“我们的船都已经扬起帆了。但要找到对临床试验的支持,真的太难了,更不用是儿童的临床试验了,哪怕我们知道他们会是最终的受益者。”近日,贝内特在接受《金融时报》的采访时回忆说。
一停滞就是几年过去。2005年,凯瑟琳·海伊(Katherine High)向贝内特伸出了橄榄枝,改变了这一僵局。海伊研究血友病,同样遭受基因治疗低谷期的困境,她跳槽至美国费城儿童医院,希望能继续开展基因治疗的临床试验。
费城儿童医院被海伊说服了,但设置了前提条件:除了研究血友病,还需要研究一个儿童病。正是这个条件,使得海伊敲开了贝内特办公室的大门。

凯瑟琳·海伊(Katherine High)
在贝内特夫妇看来,尽管有历史的前车之鉴,莱伯先天性黑蒙症的临床试验有科学理论加持的安全性。在人人捏把汗的细胞因子风暴上,莱伯先天性黑蒙症有自己的优势:眼睛是免疫豁免部位,而且体积较小,出现免疫排斥的可能性大大降低。再者,Lancelot的成功已经带来动物模型上的借鉴,犬类的眼睛大小和人类相近。
几经波折后,2008年,《新英格兰医学杂志》同时发表了来自两个独立研究团队的临床试验报告。英国伦敦大学学院和贝内特团队各自在3位LCA2患者身上进行了基因治疗,并分别汇报了术后12个月和5个月的情况。令人欣喜的是,患者都没有出现严重的免疫排斥反应,且视力都得到改善。
通过研究人员公开的手术视频,我们得以知道医生如何通过一根极细的针,将载有数以亿计的RPE65基因(cDNA)的AAV病毒注射在视网膜下腔。注射的精确位置在视网膜色素上皮细胞和脉络膜之间,这是视网膜细胞的入口。
“我们的眼球相当于照相机的镜头,眼睛后面的视网膜是屏幕。针插入的时候,它不是把‘镜头’给刺穿了,眼球上面有一层薄膜,它就贴着薄膜,转到眼睛的后面去。”祁鸣解释说:“AAV载着正常的基因拷贝,注射到那去,正常的基因拷贝就会自动整合到眼球后面的视网膜细胞里,产生正常的RPE65蛋白,然后就把‘屏幕’——视网膜修复好了。”
伦敦大学学院和贝内特团队分别进行了障碍走测试。伦敦大学学院组的病人在术前需要77秒走出迷宫,期间7次碰到障碍物,2次迷失方向。术后6个月,形成显著对的是,病人仅花14秒就找到了出口,没有一次碰到障碍物。虽然贝内特团队的障碍设计有所不同,但类似的是,患者术后能显著地避让障碍物。
得到小规模的临床实验结果后,贝内特团队分别扩大了规模,先后在2007- 2012年和2013 - 2015年开始了11人和31人的临床试验,并从先前的只做单眼,开始为双眼治疗。
三期试验的31名被试者中,2位中途退出,接受治疗的29位中有27位(93%)的视力得到显著改善,21位(72%)被试者在治疗后一年,仍能通过视力测试。
后续的跟踪观察仍在继续,按照FDA的要求,贝内特团队所在的Spark Therapeutics公司需要随访这些病人的状况直至2029年。
4100万美元/对
贝内特夫妇将LCA2基因疗法的专利授权给了Spark Therapeutics,放弃了从中抽取分红。
和贝内特不同,资本圈对Spark Therapeutics的热捧使得这个2013年成立、2015年在纳斯达克上市的基因治疗公司已经估值30亿美元。但是,华尔街看好的其实是Spark Therapeutics另一款在开发中的针对血友病的基因疗法。
“Luxturna”的商业前景并非被所有人看好。对于传统药企而言,开发像莱伯先天性黑蒙症之类的罕见儿童病的药物并不是一笔划算的买卖,可能面临收支无法平衡的局面。
尽管Spark Therapeutics表示将在明年FDA正式批准后才公布定价。但市场和圈内已经做出预测:单眼治疗70万-90万美元,双眼治疗100万美元。
荷兰UniQure公司2012年在欧洲上市的全球第一款基因治疗药物Glybera同样定价100万美元/次。高调的开场却迎来悲凉的结局:4月,UniQure公司宣布,不再重新申请Glybera的销售许可,10月后将退出市场。据悉,上市之来,Glybera仅售出一次,尽管治好了脂蛋白酯酶缺乏症。
相对于脂蛋白酯酶缺乏症1/100万的发病率,同属罕见病的莱伯先天性黑蒙症的2-3/10万要相对“寻常”,但Luxturna依然逃不过价格太过昂贵、受众面小的困境。
一些可能的付费方式或能缓解天价费用。比如8月获得FDA上市批准的诺华CAR-T(Kymria)产品定价47.5万美元/位,但表示可依据疗效付费,没有效果则退还费用。
而在疗效上,Luxturna仍然没有消除所有人的质疑。尽管患者在术后都得到显著视力改善,但和健康人相比,视力仍存在差距,也可能伴随眼震之类的副作用。还有的意见认为,Luxturna带来的疗效能持续多长时间仍有待观察。
2013年,美国佛罗里达大学眼科教授威廉·豪思沃斯(William Hauswirth)参与发表一篇论文,认为和Luxturna类似的基因疗法在术后,仍然无法阻止视网膜的继续恶化。值得一提的是,豪思沃斯受雇于另一家基因治疗公司,和Spark Therapeutics存在一定竞争关系。
患者术后是不是会随着年数的增加而视力退化?尚待时间验证。但科学家指出,已经接受过治疗的患者很难再接受第二次治疗。因为免疫系统对引入过一次的AAV病毒有“记忆”功能,或有更高风险。
但不可否定的是,Luxturna的成绩单为遗传性失明乃至其他基因疾病带来治疗的希望。
祁鸣每周四会在浙江大学第一附属医院坐诊。去年他参加学术会议,听说了Luxturna对6-48岁的病人都适用。回去后,他就给气馁的病人打气:“现在已经可以治RPE65型了,从6岁就可以治,瞎了一辈子的都可以治好。虽然你的类型暂时还没有,也不要灰心,说不定过几年就有了。”










